Novel hierarchical porous carbon materials (HPCs) were fabricated via a reactive template-induced in situ hypercrosslinking procedure. The effects of carbonization conditions on the microstructure and morphology of HPC were investigated, and the adsorption of methylene blue (MB) on HPC was explored. The as-prepared HPC has a hierarchical micro-, meso- and macropore structure, which results from the overlap of hollow nanospheres possessing microporous shells and macroporous cavities. The carbonization temperature, carbonization time and carbonization heating rate played important roles in tailoring the nanostructures of HPC. The BET specific surface area and micropore specific surface area can reach 2388 m2·g−1 and 1892 m2·g−1, respectively. Benefitting from the well-developed pore structure, the MB removal efficiency can reach 99% under optimized conditions. The adsorption kinetics and thermodynamics can be well described by a pseudo-second-order model and Langmuir model, respectively. Furthermore, such adsorption is characterized by a spontaneous endothermic process.
Research Article
Structure Control of Novel Hierarchical Porous Carbon Material and its Adsorption Properties
https://doi.org/10.21203/rs.3.rs-1012074/v1
This work is licensed under a CC BY 4.0 License
You are reading this latest preprint version
Carbon material
Hierarchical porous structure
Structure control
Adsorption
Organic porous materials are superior in considerable specific surface area (SSA) and stable chemical structure and are widely applied in the fields of adsorption, phase separation, catalysis and energy storeage1–4. For organic porous materials, research on porous structures is gradually moving in two directions5. One is to conduct in-depth research on the existing porous structure to further improve its performance. For instance, the pore size distribution was adjusted for a microporous material that adsorbs carbon dioxide, as the number and size of the microporous structure determine the amount of carbon dioxide adsorption, and a matched porous structure can exhibit the maximum adsorption capacity for gas or small molecules under the same conditions6,7. The other is to combine various pore structures through chemical reactions and to make full use of different pore structures. The increasing demand for organic porous materials in emerging fields tends to require materials with various pore structures.
Hierarchically porous carbon materials (HPCs) with micropores, mesopores and macropores are widely used in emerging fields due to their diverse pore structures with high SSA and excellent chemical stability8,9. However, the traditional synthesis method of HPC is still complicated, leading to an uncertain porous structure and even making the pores easily collapse. It is urgent to find a proper synthesis method. The preparation methods of hierarchical porous carbon materials can be divided into two methods according to different synthesis methods, where one is the hard template method and the other is the soft template method. The hard template needs to synthesize a template with a predetermined structure, followed by a series of postprocessing steps (such as carbonization). Then, a hierarchical porous carbon material can be prepared after washing the template. However, the stairts of the hard template method are as follows: (1) The preprocessing of the template is difficult. For example, pretreatment of the silica template must achieve a certain grafting rate of the active groups to ensure the success of the subsequent cross-linking reaction. (2) The subsequent postprocessing is also complicated. The selection of a carbon source and proper reaction conditions can make HPC an excellent pore structure10.
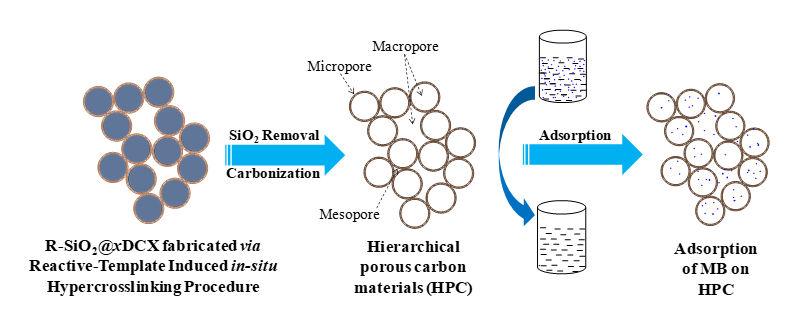
Herein, a "reactive template-induced in situ hypercrosslinking method" was successfully developed for preparing hierarchical porous polymers (HPPs) and hierarchical porous carbon materials (HPCs) using functionalized SiO2 nanospheres as the template (R-SiO2) and 1,4-p-dichlorobenzyl (DCX) as the self-crosslinking functional monomer11. The HPP and HPC prepared from this method contain a unique porous structure of micropores-mesopores-macropores in a hierarchical distribution: the microporous shell and hollow nanospheres were cross-linked and stacked with each other (Scheme 1). The abundant micro/meso/macropores are closely connected, showing a synergistic effect, which improves the properties of the materials. The effect of hypercrosslinking time on the nanostructure of HPC was studied in detail, which revealed the correlation between preparation conditions and the structural morphology of HPC. In addition, the adsorption performance of HPC toward methylene blue (MB) solution was further discussed, which provided a theoretical basis for the controllable preparation of HPC for its application in the field of dye wastewater treatment.
Preparing for reactive SiO2 nanospheres (R-SiO2) and R-SiO2@DCX
SiO2 nanospheres with an average diameter of 150 nm were synthesized through the Stöber method. SiO2 nanospheres were preprocessed to obtain reactive SiO2 nanospheres (R-SiO2). SiO2 nanospheres (0.5 g) were added to 20 ml of anhydrous tetrahydrofuran and 1.5 g of trichloro[4-(chloromethyl)phenyl]silane to a mixture with drastic sitrring. Then, 5.0 ml anhydrous tetrahydrofuran and 1.2 ml of triethylamine mixture were added under nitrogen and reacted in a nitrogen/air atmosphere for 8 and 18 hours, respectively, and then reacted in air for 18 hours. The resulting solid precipitate (named R-SiO2) was filtered and sequentially washed with ethanol and tetrahydrofuran and vacuum-dried at 60 ℃ overnight.
Generally, 0.5 g of R-SiO2 nanospheres and 1.6 g of anhydrous ferric chloride were dispersed uniformly in 10 ml dichloroethane. Then, the system was added dropwise into a solution of p-dichlorobenzene in dichloroethane at a constant speed under the protection of nitrogen after reaching 80 ℃.
Synthesis of hierarchical porous polymer (HPP) and hierarchical porous carbon material (HPC)
R-SiO2@DCX was etched with 10% HF for 24 h. The precipitate was filtered and washed with ammonia and pure water to remove the residual HF. Then, the resulting product was vacuum-dried at 60 ℃ overnight to obtain a hierarchical porous polymer (HPP). Subsequently, the HPP was carbonized at 900 ℃ in a tube furnace under the protection of a N2 atmosphere with a flow rate of 80 ml min-1 to obtain a hierarchical porous carbon material (HPC).
Characterization
Scanning electron microscopy (SEM) images were obtained on an S8010 instrument (Hitachi, Japan) at an acceleration voltage and current of 10 kV and 10 μA, respectively. The N2 adsorption-desorption isotherm of the sample was measured at 77 K by an ASAP 2460 adsorption instrument (Micromeritics, USA). The BET method was used to calculate the specific surface area, and the density functional theory (DFT) equilibrium model was used to calculate the pore size distribution. The t-plot method was used to calculate the micropore surface area and external pore surface area. An inVia laser Raman spectrometer from Renishaw Company was used to measure the microcrystalline structure of the sample. The scanning range was from 800-2000 cm-1. The excitation wavelength was 514.5 nm with an exposure time of 30 s.
The adsorption performance test
A certain amount of methylene blue (MB) solution was added to an Erlenmeyer flask, and then HPC was added. The system was kept in a water bath with magnetic field at room temperature. It was placed in a constant temperature water bath with magnetic stirring for adsorption experiments. The solution was filtered with a microporous membrane, and the concentration of the filtrate was further measured by a 722 visible spectrophotometer (Shanghai Precision Scientific Instrument Co., Ltd.). The MB adsorption rate (R) was calculated according to formula (1):

In formula (1): C0 is the concentration of MB solution before adsorption (mg·L-1); Ce is the concentration of MB solution at adsorption equilibrium (mg·L-1);
The adsorption capacity (q) was calculated according to formula (2):

In formula (2), C0 is the concentration of MB solution before adsorption (mg·L-1), Ct is the concentration of MB solution at time t (mg·L-1), V is the volume of MB solution (L), and m is the mass of HPC (g).
Characterization of the HPP
The successful preparation of R-SiO2 nanospheres was proven by a series of characterizations (Fig. S1-S3). The R-SiO2 nanospheres were further supercrosslinked in situ by means of the Friedel-Crafts reaction. As shown in Fig. 1a, the surface of the R-SiO2@DCX nanospheres presented a rough and uneven coating layer, proving the supercrosslinked reaction on the R-SiO2 surface. The unmodified SiO2 was also used to react with DCX, while polymerization was carried out only between the DCX monomer. As shown in Fig 1c, SiO2@DCX shows a flat and agglomerated surface, indicating that the cross-linking reaction did not occur. After removing the silica template (Fig. 1b), the internal cavity of the R-SiO2@DCX nanosphere maintains a complete spherical structure. The Fig. 1d showed that the average size of R-SiO2@DCX nanoarticles are at 150 nm. In addition, a series of characterizations illustrate the successful preparation of R-SiO2@DCX (Fig S4-S7).
For carbon materials, the pore structure and element formation are mainly inherient from precursors12-14. Thus, the hypercrosslinking time was changed to explore the variation in the morphology and structure of HPP to precisely control the nanostructure of HPP. Fig. 2 (SEM characterization) shows that the samples all contained structures with hollow and crossing spheres when the supercrosslinking time was continuously extended from 3 h to 24 h. As the supercrosslinking time was just 3 h (Fig. 2a), the sample showed that a small part of the spherical shells were not connected with only a small part of the crosslinking hollow spheres, and the graft layer originally covered the spherical surface was almost peeled off, leaving a few residues. As the cross-linking time was extended to 6 h (Fig. 2b), the particle size of the sample tended to be uniform, while agglomeration still occurred, and hollow spheres were almost formed with a complete appearance. When the time reached 24 hours (Fig. 2e), the hollow nanosphere shells were well stacked and regularly arranged together, forming a hollow hole-like structure with good morphology. The oligomer on the surface of the nanospheres gradually decreased with the extension of the supercrosslinking time, which was caused by the gradual consumption of the polymerized part, leading to a more complete reaction 15.
As shown in Fig. 3, all the samples presented a typical hierarchical porous structure that included micropores, mesopores and macropores16,17. The pore size distribution of HPP-Xs based on DFT calculations (Fig. 3b) showed peaks at 0.67 nm and 1.2 nm, at 27 nm and at 50-100 nm, corresponding to micropores, mesopores and macropores, respectively. The micropores were derived from the methylene bridge voids of the cross-linking between the nanospheres. As the Friedel-Crafts reaction was rapid, once the reaction occurred, it quickly polymerized to form a methylene cross-linked bridge to form a microporous structure. The changes in the supercrosslinking reaction times deeply affected the crosslinking degree of the samples18.
Table 1 summarizes the DTF calculation results based on the nitrogen absorption/desorption tests. As the reaction time was extended, the SSA and Vmicro of the samples increased 19. HPP-3 showed an SSA of 386 m2·g-1 and a Vmicro of 0.04 cm3 g-1. As the crosslinking time of HPP-3 was shortened, the reaction could not proceed completely, leading to meso/macropores occluding the majority of the pore structure. With the extension of the cross-linking time, the SSA and the Vmicro of HPP-6, HPP-12, HPP-18 and HPP-24 showed an increasing trend. The maximal SSA and Vmicro were reached when the crosslinking time increased to 24 h, where Vtotal increased from 0.04 to 0.88 cm3 g-1 and the SSA increased from 386 m2 g-1 to more than double (889 m2 g-1). The results indicated that prolonging the super-crosslinking time was helpful to the Friedel-Crafts reaction, and the degree of cross-linking rose rapidly when the time reached 18 h. Comparing the value of Vmicro, it can be found that almost all the increase in the specific surface area came from the increase in the number of micropores. Since the microporous structure in the sample was mainly derived from the self-crosslinking reaction of the benzyl group between the benzene rings, the greater the degree of crosslinking between the spheres, the denser the cross-linked bridge structure, thus forming more micropores20,21. In conclusion, HPP-24 contained the highest SSAs and the largest number of micropores, which would be the best choice to be the precursor of HPC.
Table 1
Pore structure parameters of HPP at various hypercrosslinking times
|
Sample (h) |
SBET (m2g-1) |
Smic (m2g-1) |
Sext (m2g-1) |
Vmic (cm3g-1) |
Vext (cm3g-1) |
Vtotal (cm3g-1) |
|
3 |
386 |
135 |
251 |
0.04 |
0.26 |
0.30 |
|
6 |
397 |
143 |
201 |
0.04 |
0.27 |
0.31 |
|
12 |
434 |
186 |
218 |
0.06 |
0.31 |
0.37 |
|
18 |
450 |
229 |
221 |
0.09 |
0.33 |
0.42 |
|
24 |
889 |
567 |
332 |
0.23 |
0.65 |
0.88 |
The structure characterization of HPC
Fig. 4a shows an SEM image of a typical HPC, displaying a three-dimensional nanonetwork structure with mesopores and macropores, which was formed by the stack of carbon nanospheres. The broken nanospheres possessed a hollow structure, indicating successful etching of the SiO2 template. Fig. 4b show the Raman spectrum of HPC. The characteristic peak at 1345 cm-1 was the D peak, and the G peak was at 1590 cm-1, where the D peak corresponded to the vibration of carbon atoms and the G peak was the characterization of the graphitized structure, indicating that the carbon skeleton of HPC contained a graphite-like microcrystalline structure22. Fig. 4c illustrates the N2 adsorption-desorption isotherm of HPC. In the low-pressure region (P0/P < 0.1), the adsorption isotherm increased, indicating the microporous structure of HPC, while the isotherm showed a hysteresis loop area in the medium-pressure region, indicating the existence of mesopores; when the relative pressure was close to 1.0, the adsorption capacity increased significantly without the adsorption platform, showing the presence of macropores23. These conclusions were further proven by the pore size distribution curve, which was calculated from the DFT method. As shown in Fig. 4d, the pore size was distributed from the micropores (pore size below 2 nm) and the mesopores (pores from 2-50 nm) to the macropores (pores above 50 nm), indicating a hierarchical structure.
The influence of carbonization temperature on the HPC nanostructure and SSA was further studied. Fig. 5 shows the SEM image of HPC prepared at different carbonization temperatures. This result showed that the special three-dimensional structure, which was formed by stacking hollow nanospheres, can be prepared at all carbonization temperature ranges, indicating that HPC presents good thermal stability and framework strength. Table S1 summarizes the pore structure parameters of HPC prepared under different carbonization conditions. The optimization conditions were a carbonization temperature of 1000 ℃ with a 3 h carbonization time and a carbonization heating rate of 5 ℃ min-1. The as-prepared HPC presented a high SSA of 2388 m2·g-1 and an outstanding hierarchically porous structure.
Adsorption performance of HPC
The results above showed that HPCs were microporous materials with abundant adsorption active sites. In addition, HPCs are hierarchically porous materials with connected pores, which could shorten the transmission distance between the pores, making HPCs a promising adsorbent with high-efficiency adsorption performance24. HPC-1000 with an SSA of 2388 m2 g-1 was selected as the adsorbent, and methylene blue (MB) was selected as the adsorbate to investigate the adsorption performance of HPCs. The mechanism of adsorption was discussed based on the model of adsorption kinetics, adsorption isotherm and adsorption thermodynamics.
Fig. 6 shows the adsorption kinetic curve of HPC-1000 in aqueous solution toward MB. When the adsorption time was 5 minutes, the adsorption rate reached 59%, increased to 64% and 99% at 10 minutes and 60 minutes, respectively, and then did not change substantially as the adsorption time increased, indicating that the adsorption toward MB had reached equilibrium at this time. In the early stage of adsorption, the microporous structure of HPC-1000 has a large number of active sites, leading to a high initial rate of adsorption. As the adsorption time increased, the active sites inside HPC-1000 were filled with MB molecules, resulting in adsorption equilibrium after 60 minutes. Fig. 6 illustrates a digital photo of HPC-1000 adsorbing toward MB in aqueous solution. As the time increased, the color of the MB solution became lighter and was completely colorless after 2 h, indicating the effective adsorption performance of HPC-1000 toward the MB solution.
Fig. 6 also shows the fitting results of the adsorption process using the pseudo-first-order kinetic model, pseudo-second-order kinetic model, and Weber-Morris intraparticle diffusion model25. Table 2 summarized the adsorption kinetic parameters. The correlation coefficient value (R2) of pseudo-second-order kinetics was higher with R2 values greater than 0.99. The equilibrium adsorption capacity (qe-exp) obtained in the experiment was very close to the theoretical adsorption capacity (qe-cal) calculated by the pseudo-second-order kinetic model, indicating that the pseudo-second-order kinetic model was more suitable for describing the adsorption process of HPC toward MB solution. The intraparticle diffusion model was used by the segmentation method to fit the adsorption kinetics data (Fig. 6f). As shown in Table 2, the linear correlation of the segmentation fitting was better (the R2 value can reach 0.9774), indicating that MB was inside the HPC. The diffusion could be divided into two stages, while the first stage was the rapid diffusion of MB in macropores and mesopores, and the second stage was the slow diffusion in micropores. Fig. 6 b and c show the fitting results of the Langmuir model and Freundlich model26 of the thermodynamics of HPC adsorption toward MB solution. The fitting results are summarized in Table 3, where the linear correlation coefficients of the Langmuir model are all higher than those of the Freundlich model, indicating that the adsorption process is more according to the Langmuir isotherm model, which means the main monolayer adsorption of the adsorption process27. The study of the HPC adsorption thermodynamics toward MB solution was further explored. According to the influence of different temperatures on the adsorption in the range of 298 to 318 K, the adsorption thermodynamic constants can be calculated. As shown in Table 4, the Gibbs free energy ∆G value was less than zero at the experimental temperature. As the temperature rises, the absolute value of ∆G increases, indicating that HPC adsorption toward MB solution was a spontaneous behavior. The enthalpy change value (∆H) was more than 0, indicating that the adsorption of MB by HPC is an endothermic process. The entropy change value (ΔS) reflected the degree of disorder of the solid-liquid interface, and the value ΔS>0 indicated that the adsorption process increased the degree of disorder of the molecular motion between the solid-liquid interface, indicating that HPC had a strong affinity toward MB solution28.
Table 2
Kinetic parameters at various MB concentrations
|
Kinetic equation |
C0 (mg·L-1) |
qe-exp (mg·g-1) |
Kinetic parameters |
|
Pseudo-first-order ln(qe-qt) = lnqe-K1t |
10 20 30 |
31.5 32.3 29.2 |
K1 = 0.07266, qe-cal =35.5, R2 = 0.9353 K1 = 0.07159, qe-cal =35.1, R2 = 0.9285 K1 = 0.06710, qe-cal =31.0, R2 = 0.9750 |
|
Pseudo-second-order t/qt = 1/(K2qe2)+t/qe |
10 20 30 |
48.1 57.9 77.8 |
K2 = 0.00279, qe-cal =51.8, R2 = 0.9956 K2 = 0.00337, qe-cal =59.0, R2 = 0.9966 K2 = 0.00439, qe-cal =78.2, R2 = 0.9991 |
|
Intraparticle diffusion qt = Kid t1/2 + C |
10
20
30 |
/ |
Kid1 = 3.71, C1= 19.1, R21 = 0.9774 Kid2 = 0.11, C2= 46.8, R22 = 0.9983 Kid1 = 3.53, C1= 28.4, R21 = 0.9619 Kid2 = 0.17, C2= 54.1, R22 = 0.9054 Kid1 = 2.89, C1= 53.7, R21 = 0.9505 Kid2 = 0.17, C2= 73.8, R22 = 0.9486 |
Table. 3
Langmuir and Freundlich isothermal equation parameters
|
Temperature (℃) |
Langmuir |
Frenndlich |
||||
|
qm(mg·g-1) |
KL(L·mg-1) |
R2 |
|
KF(mg·g-1) |
R2 |
|
|
25 |
125.1 |
0.0522 |
0.94996 |
|
9.0346 |
0.92111 |
|
35 |
121.1 |
0.058 |
0.99316 |
|
10.8137 |
0.97651 |
|
45 |
123.2 |
0.0588 |
0.99726 |
|
10.305 |
0.96459 |
Table. 4
Adsorption thermodynamic parameters
|
Temperature(K) |
thermodynamic parameters |
||
|
∆G(kJ·mol-1) |
∆H(kJ·mol-1) |
∆S(J·mol-1·K-1) |
|
|
298 |
-4.86 |
|
|
|
308 |
-5.34 |
14.910 |
41.947 |
|
318 |
-5.70 |
|
|
In conclusion, this paper provides a novel "reactive template-induced in situ hypercrosslinking method" to synthesize hierarchical porous carbon materials (HPCs). The effect of carbonization conditions on the nanostructure and morphology of HPC was deeply studied. The results show that the shell-macroporous hollow nanospheres were stacked on each other to form meso/macropores, forming a three-dimensional nanonetwork structure. The diameter of the CNPs was approximately 150 nm, presenting unique micropore-mesopore-macropore hierarchical distribution pore structure characteristics; by controlling the carbonization conditions, the pore structure of HPC could be effectively customized. The adsorption rate of HPC toward methylene blue (MB) small molecules in aqueous solution could reach more than 99%. In addition, the adsorption kinetics and thermodynamics toward MB were in line with the pseudosecondary adsorption kinetics model and Langmuir model. The adsorption process was homogeneous monolayer adsorption, and it was a spontaneous endothermic process.
Acknowledge:
The authors thank the Natural Science Foundation of Fujian Province, China, for financial support (Grant No. 2019J01808; 2021J011107)
- Zhang, F., Wang, K. X., Li, G. D., Chen, J. S. Hierarchical porous carbon derived from rice straw for lithium-ion batteries with high-rate performance. Electrochem. Commun. 11, 130-133 (2009).
- Jones, B. H., Lodge, T. P. Nanocasting nanoporous inorganic and organic materials from polymeric bicontinuous microemulsion templates. Polym. J. 44, 131-146 (2012).
- Zheng, Y. H. et al. Near-infrared-excited multicolor afterglow in carbon dots-based room-temperature phosphorescent materials. Angew. Chem. 133, 22427-22433 (2021).
- Meng, J. S. et al. Universal approach to fabricating graphene-supported single-atom catalysts from doped ZnO solid solution. ACS Cent. Sci. 6, 1431-1440 (2020).
- Dorin, R. M., Sai, H., Wiesner, U. Hierarchically porous materials from block copolymers. Chem. Mater. 26, 339-347 (2014).
- Ji, X. Y. et al. Capturing functional two-dimensional nanosheets from sandwich-structure vermiculite for cancer theranostics. Nat. Commun. 12, 1124 (2021).
- Zhou, H. W. et al. Robust and sensitive pressure/strain sensors from solution processable composite hydrogels enhanced by hollow-structured conducting polymers. Chem. Eng. J. 403, 126307 (2021).
- Ouyang, J. et al. In situ sprayed NIR-responsive, analgesic black phosphorus-based gel for diabetic ulcer treatment. P. Natl. Acad. Sci. USA(PNAS) 117, 28667-28677 (2020).
- Wu, J. L., Xu, F., Li, S. M., Ma, P. W., Zhang, X. C., Liu, Q. H., Fu, R. W., Wu, D. C. Porous polymers as multifunctional material platforms toward task‐specific applications. Adv. Mater. 31, 1802922 (2019).
- Wei, D. S. et al. Breaking down the intracellular redox balance with diselenium nanoparticles for maximizing chemotherapy efficacy on patient-derived xenograft models. ACS Nano 14, 16984-16996 (2020).
- Cai, L. F., Chen, L. Y., Liang, Y. R., Lu, Z. T., Xu, F., Fu, R. W., Wu, D. C. Reactive-template induced in situ hypercrosslinking procedure to hierarchical porous polymer and carbon materials. Acta Chim. Sinica 73, 600-604 (2015).
- Choi, M., Ryoo, R. Mesoporous carbons with KOH activated framework and their hydrogen adsorption. J. Mater. Chem. 17, 4204-4209 (2007).
- Zhu, Y. W., Murali, S., Stoller, M. D., Ganesh, K. J. Carbon-based supercapacitors produced by activation of graphene. Science 332, 1537-1541 (2011).
- Zhao, L., Fan, L. Z., Zhou, M. Q., Guan, H. Nitrogen-containing hydrothermal carbons with superior performance in supercapacitors. Adv. Mater. 22, 5202-5206 (2010).
- Liang, Y. R., Liang, F. X., Zhong, H., Li, Z. H., Fu, R. W., Du, D. C. An advanced carbonaceous porous network for high-performance organic electrolyte supercapacitors. J. Mater. Chem. A 1, 7000-7005 (2013).
- Nishihara, H., Kyotani, T. Energy storage: templated nanocarbons for energy storage. Adv. Mater. 24, 4466-4466 (2012).
- Petkovich, N. D., Stein, A Controlling macro-and mesostructures with hierarchical porosity through combined hard and soft templating. Chem. Soc. Rev. 42, 3721-3739 (2013).
- Estevez, L., Dua, R., Bhandari, N., Ramanujapuram, A., Wang, P., Giannelis, E. P. A facile approach for the synthesis of monolithic hierarchical porous carbons-high performance materials for amine based CO2 capture and supercapacitor electrode. Energy Environ. Sci. 6, 1785-1790 (2013).
- Yan, X. H., Xu, H., Fang, G. L., Xue, T., Meng, Z. W., Lee, J. M. A reactive template synthesis of hierarchical porous carbon and its application to supercapacitor electrodes[J]. Macromol. Mater. Eng. 305, 2000168 (2020).
- Wang, P., Qi, X. H., Zhao, W., Qian, M. Nitrogen-doped hierarchical few-layered porous carbon for efficient electrochemical energy storage. Carbon Energy 3, 349-359 (2020).
- Jung, D. S., Hwang, T. H., Lee, J. H., Koo, H. Y. Hierarchical porous carbon by ultrasonic spray pyrolysis yields stable cycling in lithium-sulfur battery. Nano Lett. 14, 4418-4425 (2014).
- Salunkhe, R. R., Kaneti, Y. V., Kim, J., Kim, J. H. Nanoarchitectures for metal-organic framework-derived nanoporous carbons toward supercapacitor applications. Acc. Chem. Res. 49, 2796-2806 (2016).
- Sing, K. S. W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 57, 603-619 (1985).
- Wang, D. W., Li, F., Liu, M., Lu, G. Q., Cheng, H. M. 3D aperiodic hierarchical porous graphitic carbon material for high-rate electrochemical capacitive energy storage. Angew. Chem. 48, 1525-1525 (2009).
- Zhang, L., Li, X., Yang, X. J., Li, Y. A rapid and selective isolation of rhodium from aqueous solution using nano-Al2O3. J. Chem. Eng. Data 57, 2647-2653 (2012).
- Zacaria, R., Gerente, C., Andres, Y., Cloirec, P. L. Adsorption of several metal ions onto a low-cost biosorbent: kinetic and equilibrium studies. Environ. Sci. Technol. 36, 2067-2073 (2002).
- Zhang, Y., Jing, L. Y., He, X. H., Li, Y. F., Ma, X. Sorption enhancement of TBBPA from water by fly ash-supported nanostructured γ-MnO2. J. Ind. Eng. Chem. 21, 610-619 (2015).
- Liu, Q. S., Zheng, T., Li, N., Wang, P., Abulikemu, G. Modification of bamboo-based activated carbon using microwave radiation and its effects on the adsorption of methylene blue. Appl. Surf. Sci. 256, 3309-3315 (2010).
No competing interests reported.
- supplimentarymaterial.docx
- SCheme1.png
Schematic of novel hierarchical porous carbon material (HPC) and methylene blue (MB) adsorption
{kind=link}