The integration of aggregation-induced emission (AIE) and two-photon absorption (2PA) into one molecule is very important because such combination would endow the material with synergetic effect for diverse and broader applications. Herein, two regioisomers based on perylene diimide and anthracene moieties are designed and synthesized. The as-prepared compounds exhibit distinct AIE and 2PA properties, where the ortho-substituted one not only exhibits enhanced and accelerated AIE performance, but also displays improved 2PA properties. This research would provide a general approach to explore efficient molecules by the marriage of AIE and 2PA that could improve their performance in various areas.
Article
Simultaneously Enhancing Aggregation-Induced Emission and Boosting Two-Photon Absorption Performance of Perylene Diimides through Regioisomerization
https://doi.org/10.21203/rs.3.rs-1027693/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 01 Jan, 2022
Read the published version in Journal of Materials Chemistry C →
Version 1
posted
You are reading this latest preprint version
aggregation-induced emission (AIE)
two-photon absorption (2PA)
regioisomers
Organic molecules exhibiting two-photon absorption (2PA) activity are of interest for a wide range of applications, including two-photon fluorescence microscopy and biological imaging,1–4 two-photon photodynamic therapy,5–6 three-dimensional (3D) microfabrication,7–8 3D optical data storage,9–10 optical power limiting,11–12 and up-converted lasing.13 To facilitate these applications, the design and synthesis of efficient 2PA dyes are essential. Recently, this field has been comprehensively reviewed.14–16 However, 2PA dyes often show low emission efficiency (fluorescence quantum yields, ΦF) when they are in high concentration or in the aggregated state, which is known as aggregation-caused quenching (ACQ). Diametrically opposite to ACQ characteristic is the abnormal fluorescence enhancement phenomenon after molecular aggregation. Tang and coworkers first coined this concept of aggregation-induced emission (AIE) in 2001 based on the study of 1-methyl-1,2,3,4,5-pentaphenylsilole.17 To date, a series of AIE molecules have been developed, including tetraphenylethenes,18–19 siloles,20–21 phosphindoles,22 etc. These AIE molecules are designed as propeller-shaped molecules because the restriction of intramolecular rotations blocks the nonradiative pathway and opens up the radiative channel when aggregated. This design strategy of AIE molecule is opposite to that of 2PA molecule, where planar structure with elongated π-conjugation favors large 2PA cross-section.23–26 Therefore, it is very important to find the right balance between optical properties and structures for optimized 2PA and AIE molecules. A special molecular design for a 2PA molecule is required not only to ensure a large two-photon activity, but also to overcome fluorescence quenching at a high concentration or in an aggregated state.
The key factor determining the AIE property is the moieties that can undergo active intramolecular motions to sufficiently dissipate excited-state energy in the isolated state.27 Thus, it is envisaged that the integration of the motional elements into the 2PA molecule may work as an effective rationale to eradicate the ACQ effect of the 2PA molecule and meanwhile retain the two-photon activity.28 Tian, Hua and coworkers reported 2PA-active multibranched molecules incorporating triazine core and triarylamine branch, in which the rotational motions of the phenyl rings aid in dissipating excited-state energy in the solution state and assist in restricting intramolecular rotation (RIR) in the aggregated state.29 Xu, Chi and coworkers reported D-π-D type 2PA molecules incorporating fluorenyl-substituted triphenylamine as electron donor and distyrylbenzene/9,10-distyrylanthracene as π-conjugated bridge, in which tetraphenylethylene is also used by non-conjugation attachment to ensure fluorescence enhancement from isolated state to aggregated state.30
The above strategy for ACQ-to-AIE transformation is taking advantage of the features of AIE units, where the number of AIE units strongly affects the AIE performance. In 2020, Tang, Jiang and coworkers reported a series of core-dendron molecules with AIE characteristics. With increasing the number of spirobifluorene dendrons, these molecules show more prominent AIE features and higher fluorescence quantum yields.31 In 2020, Tang, Ding and coworkers reported D-A structured molecules with methoxy-substituent TPE as the donor and thienopyrazine as the acceptor. With the number of the TPE increasing from two to four, the AIE amplitude also increases accordingly.32 In addition to the number of the AIE units, the substitution position is also an important factor relevant to the conversion process. Li and coworkers reported three bis[(anthracenyl)vinyl]benzene, of which ACQ is observed when the two anthracenyl groups are substituted at the meta-position of the benzene moiety. In contrast, AIE is observed when the two anthracenyl groups are substituted at the ortho-position of the benzene moiety.33 Konishi and coworkers reported a series of regioisomers of bis(piperidyl)anthracenes. The para-substituted 1,4-BPA and 9,10-BPA exhibited obviously different AIE behaviors.34 In 2020, Tang, Lu, Qian and coworkers reported two D-A-D type 2PA molecule containing donor triphenylamine and acceptor dithienobenzophenazine., where by shifting triphenylamine from the end to the bay position of dithienobenzophenazine, ACQ-to-AIE transformation is also realized.35
These successes triggered the researchers' interest in converting larger π-conjugated systems from ACQ to AIE molecules. Perylene-3,4,9,10-tetracarboxylic acid diimides (perylene diimides, PDIs) are one class of the most explored organic fluorescent materials because of their high fluorescence quantum yields in dilute solvents. However, their aggregates display drastic fluorescence quenching due to effective intermolecular π-π stacking. Therefore, the conversion of PDIs from ACQ to AIE molecules is of great significance. Up to now, there are only a few reports on the conversion of PDIs from ACQ to AIE. Tang, Gao and coworkers reported the ACQ-to-AIE transformation by modifying the bay area of PDI core with triphenylethylene.36 Tang, Sun and coworkers compared the fluorescent behaviors of PDIs substituted with one and two tetraphenylethene moieties at the bay area. The di-TPE substituted PDI exhibits better AIE performance than the mono-TPE substituted PDI.37 They also compared the AIE performance of PDIs decorated with two tetraphenylethene moieties at the bay area (1,6-/1,7-positions). These two regioisomers exhibit slightly different AIE behaviors, where 1,7-isomer shows an enhanced AIE effect compared to 1,6-isomer.38 In 2018, Li, Li, Qian and coworkers developed two PDIs (DCzPDI and SCzPDI) substituted with one and two carbazolyl moieties at the bay area, where DCzPDI exhibits a faster AIE process and greater AIE amplitude.39 To the best of our knowledge, the enhancement of AIE performance by changing the substitution position of PDI core from bay to ortho area has not been reported yet.
Molecular Design Strategy, UV-Vis Absorption and Fluorescence Emission Properties
In general, PDIs can be chemically modified in the imide-region, bay-region, or ortho-region. The substituents at the imide-region are primarily used to improve the solubility of PDIs in various solvents while minimally altering the optical and electronic properties. The modifications in the bay-region result in a twist of the two naphthalene half units in PDIs, which can tune the optical and electronic properties. The functionalization in the ortho-region can tune the optical and electronic properties without affecting the planarity of the perylene core.40 PDI is a typical electron acceptor. Thus, the attachment of an electron donor to the bay- or ortho-region of PDI core can effectively induce an intramolecular charge-transfer and finely tune the 2PA properties.
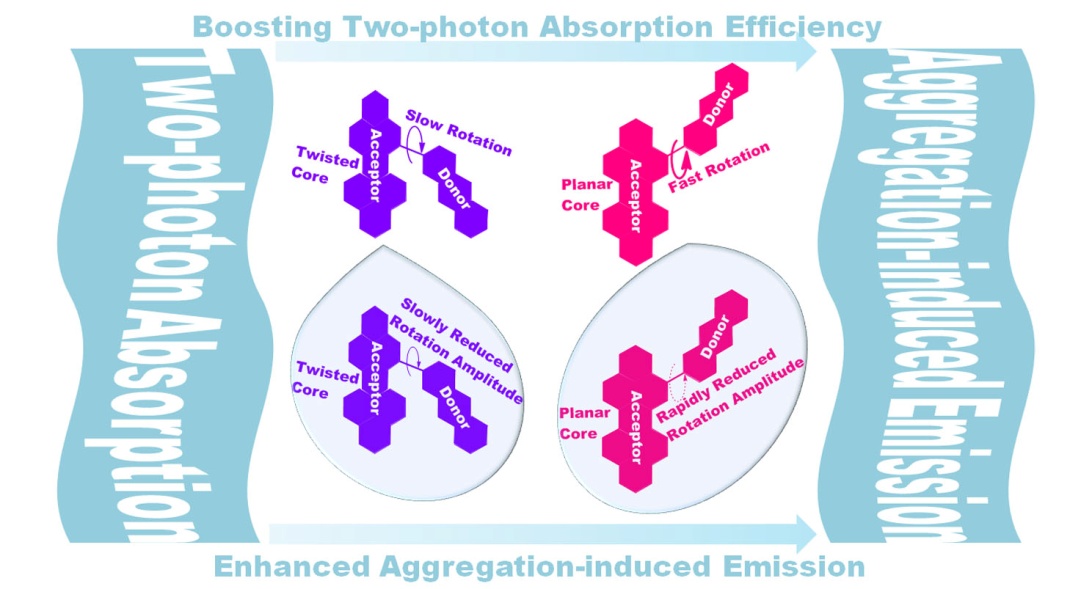
The selection of electron donor is based on two main points. Firstly, the intramolecular rotation (IMR) of donor "rotor" against the PDI core "stator" can deactivate the excited state of the molecule. For the largely planar PDI "stator", phenyl or biphenyl "rotors" are not enough to convert the emission behavior from ACQ to AIE due to the small volume.39 Therefore, a donor "rotor" with a larger size should be considered. At the same time, to avoid a big rotational barrier, the molecular weight of "rotor" should not be too large. Secondly, it is recognized that a good conjugation between building blocks as well as conformational rigidity are favored for 2PA properties. So, the donor "rotor" should effectively prolong the molecular conjugation without introducing more rotatable aromatic rings. As one kind of polycyclic aromatic hydrocarbons (PAH), anthracene (ANT) is selected as the "rotor" because of the above reasons. Herein, PDI substituted with ANT at bay- or ortho-position (B-PDI-ANT and O-PDI-ANT) have been designed and synthesized, and their structures are shown in Figure 2a.
For comparison, the UV-vis absorption spectra of PDI, B-PDI-ANT and O-PDI-ANT dyes in their dilute tetrahydrofuran (THF) solution (10−5 M) were measured and the results are displayed in Figure 2b. O-PDI-ANT and PDI show very similar absorption features: the maximum absorption bands range between 400 and 550 nm with a series of well-resolved vibronic structures. This spectral feature indicates that the ortho-substitution by ANT has little influence on the planarity of the PDI core. The above results are quite different from that observed for the PDI dye with a substituent at bay-position. Due to the steric hindrance, the perylene core is twisted in bay-substituted PDI dye. In comparison with the spectrum of O-PDI-ANT, the line-shape of the spectrum for the B-PDI-ANT is broader and displays less vibronic structures due to the loss of the planarity of the perylene core. According to the literature, the symmetrically forbidden S0-S2 transition band of bay-substituted PDI is enhanced due to the core-twisting induced broken symmetry.41 In this case, the spectral features of B-PDI-ANT are typical of PDI derivatives with modification at the bay-positions.
The fluorescence emission spectra of PDI, B-PDI-ANT and O-PDI-ANT dyes in THF solution (10−6 M) are shown in Figure 2(c). Upon excitation at 525 nm in THF, PDI displays fluorescence emission with a peak and a pronounced shoulder at 530 nm and 571 nm, respectively. The emission and absorption spectra of PDI display a mirror image relationship, denoting that no aggregation occurs at this concentration in THF at room temperature.42 The fluorescence quantum yields (ΦF) of PDI in THF solution is 70% using rhodamine B (ΦF = 97% in ethanol) as the calibration. At the same condition, B-PDI-ANT and O-PDI-ANT show significantly red-shifted emission bands and drastically reduced emission efficiencies (ΦF = 0.9% and 0.1%, respectively). The fluorescence red-shift effect might be attributed to the expansion of the π-conjugated system by introducing ANT to the PDI core (bay- or ortho-position). The fluorescence quenching effect may be due to the intramolecular charge-transfer between ANT and PDI moieties, which is frequently observed in the D-π-A systems constructed by one electron-donating unit and one electron-withdrawing unit. Also, upon UV illumination, we noted that the wet spots of both B-PDI-ANT and O-PDI-ANT on TLC plates were nonemissive. However, they became emissive when they became dried spots. This result implies that both B-PDI-ANT and O-PDI-ANT would be AIE-active. Thus, the fluorescence quenching of B-PDI-ANT and O-PDI-ANT in their dilute THF solutions can be tentatively associated with the intramolecular rotations of the ANT groups, which could exhaust the energy of the excited state.
AIE Properties
To verify the AIE property, fluorescence emission spectra were tested to monitor the emission intensity fluctuation of the molecules in THF-water mixtures at various water volume fractions (fw). As shown in Figure 3a and 3c, the emission intensities are intensified for these two molecules when fw increases from 0 to 90%, exhibiting an AIE behavior. The comparison of their characteristic AIE properties discloses important information. Firstly, the evolution of the AIE process for their aggregates is different. To have a quantitative picture of their AIE process, we estimated their ΦF values in THF-water mixtures, using rhodamine B as the reference. As shown in Figure 3b and 3d, for B-PDI-ANT and O-PDI-ANT, the ΦF of the THF solution are very low (ΦF=0.93% and 0.13%) and are almost unchanged when water is added up to 50%, but start to increase swiftly upon addition of water to 80% and 50%, respectively. The trajectory of the ΦF change for B-PDI-ANT suggests that the molecules start to aggregate at a water fraction of >50%, but the highly twisted PDI core might provide enough room for the intramolecular rotation (IMR) of the ANT in B-PDI-ANT. Thus, the channel for consuming the excitation energy cannot effectively be switched off. As a result, B-PDI-ANT shows subdued emission at the fw of 60-80%. However, only when the molecules are packed much more tightly at the fw>80%, the aggregation-induced emission would be realized. A similar situation has also been reported by previous literature.43 Unlike B-PDI-ANT, O-PDI-ANT exhibits accelerated AIE behavior. When the fw reaches 50%, aggregation occurs, resulting in a rapid increase in the ΦF based on the restriction of the intramolecular rotation (RIR) mechanism. The planar conformation of the PDI core could effectively diminish the molecular rotation to improve the efficiency of the radiative process. Accompanying the further increase of fw to 70%, the ΦF decreases, probably due to the transformation from ordered to random aggregates. Secondly, the degree of the AIE enhancement for their aggregates is different. Their aggregates formed in THF-water mixtures (fw = 60%) display increased emission with the ΦF values of 2.8 and 3.9%, respectively, resulting in the AIE amplification factors (αAIE = ΦF,aggr/ΦF,soln) of 3 and 30, respectively. Their highest ΦF values are achieved at fw of 90%, with the αAIE of 18 and 34, respectively. These data suggest that O-PDI-ANT has a better AIE performance than B-PDI-ANT.
Femtosecond Transient Absorption and Theoretical Calculation
In order to study the excited state dynamics and especially the AIE mechanism of B-PDI-ANT and O-PDI-ANT, the femtosecond transient absorption (fs-TA) spectroscopy studied on PDI dyads were performed. The pump wavelength was 525 nm. Figure S5-S6 and Figure 4a show the fs-TA spectra of B-PDI-ANT. As shown in Figure S5, at the earlier delay time from 0 to 0.39 ps, the intensity of the broad band (465-625 nm) with centered wavelength at 528 nm increases, which is attributed to the ground-state bleaching (GSB). Two bands at short wavelength (centered at 350 and 420 nm, respectively) and one band at long wavelength (centered at 680 nm) also increase at this delay time range, which are attributed to the excited-state absorption (ESA) associated with the S0→S1. As shown in Figure S6, at later delay time from 0.39 to 6.58 ps, the ESA bands show apparent hypsochromic shift with the center wavelength emerging at 325 nm, 396 nm, and 675 nm, respectively. This process is mainly associated with the twisted intramolecular charge-transfer (TICT) from ANT to PDI groups. As shown in Figure 4a, at the last delay time from 6.58 to 925 ps, the GSB and ESA bands gradually drop to zero. The rotation of the ANT group in B-PDI-ANT is the primary channel to relax its excited state energy, which completely overwhelms the radiative process. As shown in Figure S7, global fitting analysis of the decay kinetics at all wavelengths yields three time constants: 0.43 (τ1), 1.92 (τ2) and 250 ps (τ3), which is consistent with the three excited-state decay processes observed in Figure S5-S6 and Figure 4a. O-PDI-ANT is a positional isomer to B-PDI-ANT. Nevertheless, the fs-TA spectra results show that the excited state of O-PDI-ANT undergoes a process apparently different from that of B-PDI-ANT. As shown in Figure S9, at the first stage (0-0.36 ps), the GSB band (450-550 nm) is not a broad peak but multiple peaks with fine structure (457, 488, and 528 nm), which is in agreement with the steady-state absorption spectrum. At the same time, a stimulated emission (SE) band is observed at 572 nm, which corresponds to the steady-state fluorescence emission. As shown in Figure S10, at the second stage (0.36-2.87 ps), the decrease of the SE peak at 572 nm is seen, accompanied by the blue-shift of the ESA peaks at 335, 402 and 678 nm. This reveals the conversion from fluorescence emission to ICT. Therefore, the excited state of O-PDI-ANT is mainly consumed by the nonradiative process rather than the radiative process. As shown in Figure 4b, in the third stage (2.87-7510 ps), the GSB and ESA bands completely disappears, which is attributed to the rotation of the ANT group in O-PDI-ANT. As shown in Figure S11, global fitting of the kinetics for O-PDI-ANT also obtains three-time constants: 0.36 (τ1), 3.00 (τ2) and 39 ps (τ3). These values denote the procedures of excitation, TICT and rotation. The τ3 for O-PDI-ANT is significantly shorter than that of B-PDI-ANT, indicating the rotation process dramatically accelerates in O-PDI-ANT. It is worthy to note that two new positive peaks at 470 and 504 nm begin to increase at this stage, suggesting an alternative relaxed pathway for O-PDI-ANT. An intersystem crossing may take place to generate a triplet state. The formation of the triplet state of O-PDI-ANT was confirmed with nanosecond transient absorption (ns-TA) spectroscopy (Figure S13-S14). The results and discussion are available in the ESI†.
Next, fs-TA spectra (Figure S16-S27) of B-PDI-ANT and O-PDI-ANT were recorded on their aggregated states in THF and H2O of different proportions. An increase in the proportion of H2O leads to the eventual aggregation of molecules, and hence, a corresponding change is observed in the fs-TA spectra. The study above shows that the ΦF of B-PDI-ANT and O-PDI-ANT do not show an obvious change until the proportion of H2O increase to 60%. Thus, the measurements were conducted at the fw = 60 %. For B-PDI-ANT, the measurement is also conducted at the fw = 90 % because a further significant increase in the ΦF of B-PDI-ANT is observed at the fw = 90 %. It can be easily seen that the GSB and ESA bands show an apparent redshift upon aggregation. To quantitatively describe the difference, the decay dynamics are fitted at the selected wavelength. In comparison to the global fitting results, the similar time constants of B-PDI-ANT and O-PDI-ANT can be obtained by using the wavelength fitting at 430 nm. Therefore, these two wavelengths are selected. As shown in Figure S28, for non-aggregated B-PDI-ANT in THF, the rotation amplitude is 38.5%, but this value shows a slight decrease to 31.3% upon aggregation in THF/H2O (fw = 60%). A larger extent of decline (from 30.4 to 7.95%) is measured for the rotational amplitude of O-PDI-ANT between the non-aggregated state in THF and the aggregated state in THF/H2O (fw = 60%). Such a decrease in the rotational amplitude could explain the increase of the ΦF values of aggregated molecules. These results provide evidence of rapid ΦF growth for O-PDI-ANT within the aggregates as the rotation can be constrained much quicker than that of B-PDI-ANT.
To obtain a deep insight into the different ground- and excited-state properties between B-PDI-ANT and O-PDI-ANT, TD-DFT calculation using a CAM-B3LYP/6-31G(d) method based on Gaussian 16 is performed.44-45 As the crystal structures were rather challenging to obtain, the optimized molecular structures are examined at the ground state. As shown in Figure S29 and S31, the perylene core of O-PDI-ANT is more planar than that of B-PDI-ANT, as also indicated by their UV/Vis spectra. Similar results are observed in the optimized molecular structures at the excited state (Figure S30 and S32). The more twisted conformation of perylene core in B-PDI-ANT should be ascribed to the introduction of the ANT group into the bay-position of PDI. We also calculated the rotational barriers (potential energy surface, PES) of ANT group in the ground state and excited state for isolated B-PDI-ANT and O-PDI-ANT. As shown in Figure 4c and 4d, the PESs of O-PDI-ANT exhibit "single-well", but those of B-PDI-ANT display "double-well". Not surprisingly, the intramolecular rotation of the ANT group in O-PDI-ANT is free because no energy barrier exists. In contrast, the intramolecular rotation of the ANT group in B-PDI-ANT should overcome an energy barrier of 1.1-1.8 kcal/mol at the ground state and 4.1-6.4 kcal/mol at the first excited state. Overcoming these energy barriers will cause a longer lifetime for the ANT group in B-PDI-ANT to rotate. Therefore, the PES calculations provide some evidence to explain the dynamic differences of the solution-phase results. As shown in Figure S33, B-PDI-ANT and O-PDI-ANT exhibit the charge-transfer nature for the spatially separated HOMOs and LUMOs. Their HOMOs are restricted on the ANT groups, while LUMOs are distributed to the PDI core. The TICT process can be suppressed by the physical constraint in the packing mode, thus the amplitude of TICT shows obvious decrease for B-PDI-ANT (from 27.6 to 9.08%) and O-PDI-ANT (from 28.6 to 10.8%) upon aggregation in THF/H2O (fw = 60%).
Two-photon Absorption Properties
As shown in Figure 5a, the 2PA spectrum of PDI parent shows a weak absorption with a maximum 2PA cross-section of 95 GM at 800 nm, which is also consistent with previously reported 2PA data for unsubstituted PDI parents.46 In the measuring range of 800–900 nm located at the first near-infrared (NIR-I) window, B-PDI-ANT exhibits the 2PA cross-sections ranging from 404 GM at 800 nm to 332 GM at 900 nm, which are 4 and 12 folds that of parent PDI, respectively. Within the same scanned wavelength range, O-PDI-ANT exhibits a quick increase in the δ values from 117 GM at 800 nm to 2159 GM at 900 nm, which are 1 and 77 folds of parent PDI, respectively. Particularly, in the measuring range of 900–1100 nm located at the second near-infrared (NIR-II) window, B-PDI-ANT exhibits an increase in the δ values from 332 GM at 800 nm to 969 GM at 1100 nm, which is similar to the most cases of small molecules based on bay-substituted PDI. In sharp contrast to B-PDI-ANT, O-PDI-ANT shows very large δ values (2159-4787 GM) across the same measuring wavelength range of 900-1100 nm. And the maximum δ is 4787 GM at 1100 nm. The significant enhancement of the δ values for O-PDI-ANT could be attributed to its structural features: that is a more extensive conjugated system because of the planar perylene core. This structural feature of O-PDI-ANT can be beneficial to π-electronic delocalization and afford more effective coupling channels to increase δ values. We also summarized the previously reported 2PA data for small organic molecules containing one or two PDI unit in recent ten years from 2011-2021 (Figure 5b and Table S3).47-58 Herein, to make the δ values (measured under different experimental conditions) comparable, all of the δ values presented here are measured in the femtosecond regime, which can prevent the contribution from the excited-state absorption known to lead to artificially enhanced δ values when conducted in the nanosecond regime.59 It is worth mentioning that there are very few reports on ortho-substituted PDI and our result is among the highest δ values reported for small organic molecules containing one or two PDI units. And inspired by the excellent 2PA performance, this work would provide critical guidelines for the design of highly effective 2PA dyes.
In summary, we have developed a practical strategy to enhance the aggregation-induced emission (AIE) by altering the substitution position of perylene diimides (PDI) from bay to ortho area. On one hand, such a regioisomerization enhances the AIE performance. For bay-substituted product, the fluorescence quantum efficiency of the aggregates formed in THF-water mixtures (fw = 90%) is 18 folds that of the dilute THF solution. While, for ortho-substituted product, the fluorescence quantum efficiency of the aggregates formed in THF-water mixtures (fw = 90%) is 34 folds that of the dilute THF solution. Besides, the AIE process is accelerated in the case of the ortho-substituted product. The threshold of the dramatic fluorescence quantum efficiency increase appears at a water fraction of 80% for bay-substituted product, but only 50% for ortho-substituted product. The enhancement of the AIE performance is probably because of the restriction of the fast rotation process of the anthracene group in the ortho-substituted product. The acceleration of the AIE process is probably because of the rapid drop of the amplitude for the rotation process. The reasons for the enhanced and accelerated AIE are supported by the femtosecond transient absorption spectroscopy study of the bay- and ortho-substituted product in their isolated and aggregated state. Meanwhile, the theoretical calculation also proves that the rotation of anthracene group for bay-substituted product needs to overcome a small energy barrier; in contrast, the rotation of anthracene group for ortho-substituted product is free because no energy barrier exists. On the other hand, such a regioisomerization significantly improves the two-photon absorption (2PA) properties. For bay-substituted product, the maximum 2PA cross-section is 969 GM at 1100 nm, while that for ortho-substituted product is 4787 GM at the same wavelength. The large increase in the 2PA cross-section is ascribed to the improved planarity of the perylene core in the ortho-substituted product. These results demonstrate the feasibility to enhance the AIE and boost the 2PA performance by simply changing the substitution position of PDI from bay to ortho area. It is believed that, inspired by these preliminary results, many other ortho-substituted PDI with better fluorescence property and 2PA performance could be further developed.
Synthesis and characterization of the AIE molecules. All the AIE molecules were synthesized using Suzuki-coupling protocols as detailed in Supplementary Information. Chemical structures were confirmed by 1H NMR and 13C NMR spectroscopy using a Brucker-Biosipin Avance 400 MHz spectrometer in CDCl3 at room temperature.
Optical property spectroscopy. UV-Vis absorption spectra and fluorescence emission spectra were determined with a Shimadzu UV-Vis-2600 spectrophotometer and a Shimadzu RF-6000 spectrophotometer.
Transient absorption spectroscopy. The fs-TA measurements were performed based on a femtosecond Ti:sapphire regenerative amplified Ti:sapphire laser system (Coherent, Astrella) and an automated data acquisition system (Ultra-fast Systems, HeliosFire). The half pulse width of output from amplifier is 80 fs. The probe pulse was obtained by using approximately 1 W of the amplified 800 nm output from the Spitfire to generate a white-light continuum (320-780 nm) in a sapphire plate or CaF2 crystal. The maximum extent of the temporal delay was 8000 ps. The instrument response function was determined to be 200 fs. At each temporal delay, data were averaged for 2 s and collected by the acquisition system. The probe beam was split into two before passing through the sample. One probe beam traveled through the sample, the other was sent directly to the reference spectrometer that monitored the fluctuations in the probe beam intensity. Fiber optics was coupled to a multichannel spectrometer with a CMOS sensor that had a 1.5 nm intrinsic resolution. For the experiments described in this study, the sample solution was excited by a 525 nm pump beam which is obtained by using femtosecond TOPAS amplifier. The solutions were excited in a 2 mm path-length cuvette with a sample concentration of 0.2 mM throughout the data acquisition. The data were stored as three-dimensional (3D) wavelength-time-absorbance matrices that were exported for use with the fitting software. Therefore, in order to remove the contributions of the solvent, the raw spectra of the sample were subtracted by the spectra of the solvent under the same conditions.
Kinetics fitting method. Equation 1 shows the exponential function for wavelength kinetics fitting. The wavelength kinetics fitting for B-PDI-ANT and O-PDI-ANT are both fitted by three exponential functions and result in three lifetimes at 430 nm.

Potential energy surface scanning method. The potential surface energy scanning (PES) was obtained from DFT and TD-DFT calculation using Gaussian 16, employing M062X method and 6-311g (d,p) basis set with an SMD solvent model of water. The scanning mainly investigates the energy change with different dihedral between perylene ring and the anthracene ring. The step size was set to be 10 degree from -10° to -170° (The situation in 0° was excluded because the two rings were totally overlapped). The energy of the optimized geometry at ground state was set as 0, and all energy in PES used the relative energy.
Two-photon excited fluorescence spectroscopy. A homemade spectrofluorometer was used while a femtosecond pulsed laser (Chameleon Discovery NX, Coherent), with pulse duration, ≈140 fs (FWHM), 80 MHz, repetition rate, tuning range 800-1100 nm served as the excitation source.2PA measurements were performed in 10 mm spectrofluorometric quartz cuvettes with the dye concentrations of 5 × 10−5 M.Possible reabsorption of fluorescence emission was analyzed and taken into account. The quadratic dependence of two-photon induced fluorescence intensity on the excitation power was checked for each excitation wavelength. The reference is Rhodamine B with a concentration of 5 × 10−5 M solution in spectroscopic grade methanol. The samples are measured in spectroscopic grade THF. In the measuring range of excitation wavelengths, from 800 to 1100 nm, linear absorption could be neglected for all of the solutions. For comparison, the 2PA spectrum of PDI parent is also measured at the same condition.
Acknowledgements
This work is supported by National Natural Science Foundation of China (52073167, 21702132) and Guangdong Basic and Applied Basic Research Foundation (2021A1515011879).
Author Contributions
Q. Z. and L. X. conceived the project; L. X., X. L. and L. Y. synthesized the molecules. L. X. and X. L. characterized the molecules structures; Z. D. performed the theoretical calculations; S. Y. and X. L. performed the transient absorption experiments; J. W. and X. L. performed the two-photon excited fluorescence experiments; Q. Z., L. X., M. D. Li, F. L and X. F. J. discussed the experiments and results; L. X. wrote the manuscript; Q. Z., M. D. Li and L. X. revised the manuscript. All of the authors commented on the manuscript.
Competing interests
The authors declare no competing interests.
- Zipfel, W. R.; Williams, R. M.; Webb, W. W., Nonlinear magic: multiphoton microscopy in the biosciences. Nature Biotechnology 2003, 21 (11), 1369–1377.
- Helmchen, F.; Denk, W., Deep tissue two-photon microscopy. Nature Methods 2005, 2 (12), 932–940.
- Wu, L.; Liu, J.; Li, P.; Tang, B.; James, T. D., Two-photon small-molecule fluorescence-based agents for sensing, imaging, and therapy within biological systems. Chemical Society Reviews 2021, 50 (2), 702–734.
- Kim, H. M.; Cho, B. R., Small-Molecule Two-Photon Probes for Bioimaging Applications. Chemical Reviews 2015, 115 (11), 5014–5055.
- Fisher, W. G.; Partridge Jr., W. P.; Dees, C.; Wachter, E. A., Simultaneous Two-Photon Activation of Type-I Photodynamic Therapy Agents. Photochemistry and Photobiology 1997, 66 (2), 141–155.
- Shen, Y.; Shuhendler, A. J.; Ye, D.; Xu, J.-J.; Chen, H.-Y., Two-photon excitation nanoparticles for photodynamic therapy. Chemical Society Reviews 2016, 45 (24), 6725–6741.
- LaFratta, C. N.; Fourkas, J. T.; Baldacchini, T.; Farrer, R. A., Multiphoton Fabrication. Angewandte Chemie International Edition 2007, 46 (33), 6238–6258.
- Xing, J.-F.; Zheng, M.-L.; Duan, X.-M., Two-photon polymerization microfabrication of hydrogels: an advanced 3D printing technology for tissue engineering and drug delivery. Chemical Society Reviews 2015, 44 (15), 5031–5039.
- Parthenopoulos, D. A.; Rentzepis, P. M., Three-Dimensional Optical Storage Memory. Science 1989, 245 (4920), 843–845.
- Kawata, S.; Kawata, Y., Three-Dimensional Optical Data Storage Using Photochromic Materials. Chemical Reviews 2000, 100 (5), 1777–1788.
- W. Spangler, C., Recent development in the design of organic materials for optical power limiting. Journal of Materials Chemistry 1999, 9 (9), 2013–2020.
- Pascal, S.; David, S.; Andraud, C.; Maury, O., Near-infrared dyes for two-photon absorption in the short-wavelength infrared: strategies towards optical power limiting. Chemical Society Reviews 2021, 50 (11), 6613–6658.
- Lin, T.-C.; Chung, S.-J.; Kim, K.-S.; Wang, X.; He, G. S.; Swiatkiewicz, J.; Pudavar, H. E.; Prasad, P. N., Organics and Polymers with High Two-Photon Activities and their Applications. In Polymers for Photonics Applications II: Nonlinear Optical, Photorefractive and Two-Photon Absorption Polymers, Lee, K.-S., Ed. Springer Berlin Heidelberg: Berlin, Heidelberg, 2003; pp 157–193.
- Xu, L.; Lin, W.; Huang, B.; Zhang, J.; Long, X.; Zhang, W.; Zhang, Q., The design strategies and applications for organic multi-branched two-photon absorption chromophores with novel cores and branches: a recent review. Journal of Materials Chemistry C 2021, 9 (5), 1520–1536.
- Xu, L.; Zhang, J.; Yin, L.; Long, X.; Zhang, W.; Zhang, Q., Recent progress in efficient organic two-photon dyes for fluorescence imaging and photodynamic therapy. Journal of Materials Chemistry C 2020, 8 (19), 6342–6349.
- Liang, X.; Zhang, Q., Recent progress on intramolecular charge-transfer compounds as photoelectric active materials. Science China Materials 2017, 60 (11), 1093–1101.
- Luo, J.; Xie, Z.; Lam, J. W. Y.; Cheng, L.; Chen, H.; Qiu, C.; Kwok, H. S.; Zhan, X.; Liu, Y.; Zhu, D.; Tang, B. Z., Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chemical Communications 2001, (18), 1740–1741.
- Zhao, Z.; Lu, P.; Lam, J. W. Y.; Wang, Z.; Chan, C. Y. K.; Sung, H. H. Y.; Williams, I. D.; Ma, Y.; Tang, B. Z., Molecular anchors in the solid state: Restriction of intramolecular rotation boosts emission efficiency of luminogen aggregates to unity. Chemical Science 2011, 2 (4), 672–675.
- Zhao, Z.; Lam, J. W. Y.; Tang, B. Z., Tetraphenylethene: a versatile AIE building block for the construction of efficient luminescent materials for organic light-emitting diodes. Journal of Materials Chemistry 2012, 22 (45), 23726–23740.
- Zhao, Z.; He, B.; Tang, B. Z., Aggregation-induced emission of siloles. Chemical Science 2015, 6 (10), 5347–5365.
- Zhao, Z.; Wang, Z.; Lu, P.; Chan, C. Y. K.; Liu, D.; Lam, J. W. Y.; Sung, H. H. Y.; Williams, I. D.; Ma, Y.; Tang, B. Z., Structural Modulation of Solid-State Emission of 2,5-Bis(trialkylsilylethynyl)-3,4-diphenylsiloles. Angewandte Chemie International Edition 2009, 48 (41), 7608–7611.
- Bu, F.; Wang, E.; Peng, Q.; Hu, R.; Qin, A.; Zhao, Z.; Tang, B. Z., Structural and Theoretical Insights into the AIE Attributes of Phosphindole Oxide: The Balance Between Rigidity and Flexibility. Chemistry – A European Journal 2015, 21 (11), 4440–4449.
- Albota, M.; Beljonne, D.; Brédas, J.-L.; Ehrlich, J. E.; Fu, J.-Y.; Heikal, A. A.; Hess, S. E.; Kogej, T.; Levin, M. D.; Marder, S. R.; McCord-Maughon, D.; Perry, J. W.; Röckel, H.; Rumi, M.; Subramaniam, G.; Webb, W. W.; Wu, X.-L.; Xu, C., Design of Organic Molecules with Large Two-Photon Absorption Cross Sections. Science 1998, 281 (5383), 1653–1656.
- He, G. S.; Tan, L.-S.; Zheng, Q.; Prasad, P. N., Multiphoton Absorbing Materials: Molecular Designs, Characterizations, and Applications. Chemical Reviews 2008, 108 (4), 1245–1330.
- Pawlicki, M.; Collins, H. A.; Denning, R. G.; Anderson, H. L., Two-Photon Absorption and the Design of Two-Photon Dyes. Angewandte Chemie International Edition 2009, 48 (18), 3244–3266.
- Xu, L.; Zhu, H.; Long, G.; Zhao, J.; Li, D.; Ganguly, R.; Li, Y.; Xu, Q.-H.; Zhang, Q., 4-Diphenylamino-phenyl substituted pyrazine: nonlinear optical switching by protonation. Journal of Materials Chemistry C 2015, 3 (35), 9191–9196.
- Mei, J.; Leung, N. L. C.; Kwok, R. T. K.; Lam, J. W. Y.; Tang, B. Z., Aggregation-Induced Emission: Together We Shine, United We Soar! Chemical Reviews 2015, 115 (21), 11718–11940.
- Xu, S.; Duan, Y.; Liu, B., Precise Molecular Design for High-Performance Luminogens with Aggregation-Induced Emission. Advanced Materials 2020, 32 (1), 1903530.
- Jiang, Y.; Wang, Y.; Hua, J.; Tang, J.; Li, B.; Qian, S.; Tian, H., Multibranched triarylamine end-capped triazines with aggregation-induced emission and large two-photon absorption cross-sections. Chemical Communications 2010, 46 (26), 4689–4691.
- Xu, B.; He, J.; Liu, Y.; Xu, B.; Zhu, Q.; Xie, M.; Zheng, Z.; Chi, Z.; Tian, W.; Jin, C.; Zhao, F.; Zhang, Y.; Xu, J., High-performance two-photon absorption luminophores: large action cross sections, free from fluorescence quenching and tunable emission of efficient non-doped organic light-emitting diodes. Journal of Materials Chemistry C 2014, 2 (17), 3416–3428.
- Liu, D.; Wei, J. Y.; Tian, W. W.; Jiang, W.; Sun, Y. M.; Zhao, Z.; Tang, B. Z., Endowing TADF luminophors with AIE properties through adjusting flexible dendrons for highly efficient solution-processed nondoped OLEDs. Chemical Science 2020, 11 (27), 7194–7203.
- Qi, J.; Duan, X.; Cai, Y.; Jia, S.; Chen, C.; Zhao, Z.; Li, Y.; Peng, H.-Q.; Kwok, R. T. K.; Lam, J. W. Y.; Ding, D.; Tang, B. Z., Simultaneously boosting the conjugation, brightness and solubility of organic fluorophores by using AIEgens. Chemical Science 2020, 11 (32), 8438–8447.
- Li, Y.-X.; Chen, Z.; Cui, Y.; Xia, G.-M.; Yang, X.-F., Substitution Position Directing the Molecular Packing, Electronic Structure, and Aggregate Emission Property of Bis[2-(9-anthracenyl)vinyl]benzene System. The Journal of Physical Chemistry C 2012, 116 (10), 6401–6408.
- Sasaki, S.; Igawa, K.; Konishi, G.-i., The effect of regioisomerism on the solid-state fluorescence of bis(piperidyl)anthracenes: structurally simple but bright AIE luminogens. Journal of Materials Chemistry C 2015, 3 (23), 5940–5950.
- Li, Y.; Liu, S.; Ni, H.; Zhang, H.; Zhang, H.; Chuah, C.; Ma, C.; Wong, K. S.; Lam, J. W. Y.; Kwok, R. T. K.; Qian, J.; Lu, X.; Tang, B. Z., ACQ-to-AIE Transformation: Tuning Molecular Packing by Regioisomerization for Two-Photon NIR Bioimaging. Angewandte Chemie International Edition 2020, 59 (31), 12822–12826.
- Zhao, Z.; Gao, S.; Zheng, X.; Zhang, P.; Wu, W.; Kwok, R. T. K.; Xiong, Y.; Leung, N. L. C.; Chen, Y.; Gao, X.; Lam, J. W. Y.; Tang, B. Z., Rational Design of Perylenediimide-Substituted Triphenylethylene to Electron Transporting Aggregation-Induced Emission Luminogens (AIEgens) with High Mobility and Near-Infrared Emission. Advanced Functional Materials 2018, 28 (11), 1705609.
- Zhao, Q.; Zhang, X. A.; Wei, Q.; Wang, J.; Shen, X. Y.; Qin, A.; Sun, J. Z.; Tang, B. Z., Tetraphenylethene modified perylene bisimide: effect of the number of substituents on AIE performance. Chemical Communications 2012, 48 (95), 11671–11673.
- Zhao, Q.; Zhang, S.; Liu, Y.; Mei, J.; Chen, S.; Lu, P.; Qin, A.; Ma, Y.; Sun, J. Z.; Tang, B. Z., Tetraphenylethenyl-modified perylene bisimide: aggregation-induced red emission, electrochemical properties and ordered microstructures. Journal of Materials Chemistry 2012, 22 (15), 7387–7394.
- Zong, L.; Zhang, H.; Li, Y.; Gong, Y.; Li, D.; Wang, J.; Wang, Z.; Xie, Y.; Han, M.; Peng, Q.; Li, X.; Dong, J.; Qian, J.; Li, Q.; Li, Z., Tunable Aggregation-Induced Emission Nanoparticles by Varying Isolation Groups in Perylene Diimide Derivatives and Application in Three-Photon Fluorescence Bioimaging. ACS Nano 2018, 12 (9), 9532–9540.
- Sun, M.; Müllen, K.; Yin, M., Water-soluble perylenediimides: design concepts and biological applications. Chemical Society Reviews 2016, 45 (6), 1513–1528.
- Wang, Y. J.; Li, Z.; Tong, J.; Shen, X. Y.; Qin, A.; Sun, J. Z.; Tang, B. Z., The fluorescence properties and aggregation behavior of tetraphenylethene–perylenebisimide dyads. Journal of Materials Chemistry C 2015, 3 (15), 3559–3568.
- Roy, R.; Sajeev, N. R.; Sharma, V.; Koner, A. L., Aggregation Induced Emission Switching Based Ultrasensitive Ratiometric Detection of Biogenic Diamines Using a Perylenediimide-Based Smart Fluoroprobe. ACS Applied Materials & Interfaces 2019, 11 (50), 47207–47217.
- Huang, J.; Yang, X.; Wang, J.; Zhong, C.; Wang, L.; Qin, J.; Li, Z., New tetraphenylethene-based efficient blue luminophors: aggregation induced emission and partially controllable emitting color. Journal of Materials Chemistry 2012, 22 (6), 2478–2484.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox in Gaussian 16 Rev. C.01, Vol. Wallingford, CT, 2016.
- Humphrey, W.; Dalke, A.; Schulten, K., VMD: Visual molecular dynamics. Journal of Molecular Graphics 1996, 14 (1), 33–38.
- De Boni, L.; Constantino, C. J. L.; Misoguti, L.; Aroca, R. F.; Zilio, S. C.; Mendonça, C. R., Two-photon absorption in perylene derivatives. Chemical Physics Letters 2003, 371 (5), 744–749.
- Huang, C.; Sartin, M. M.; Siegel, N.; Cozzuol, M.; Zhang, Y.; Hales, J. M.; Barlow, S.; Perry, J. W.; Marder, S. R., Photo-induced charge transfer and nonlinear absorption in dyads composed of a two-photon-absorbing donor and a perylene diimide acceptor. Journal of Materials Chemistry 2011, 21 (40), 16119–16128.
- Zhang, J.; Fischer, M. K. R.; Bäuerle, P.; Goodson, T., Energy Migration in Dendritic Oligothiophene-Perylene Bisimides. The Journal of Physical Chemistry B 2013, 117 (16), 4204–4215.
- Sartin, M. M.; Huang, C.; Marshall, A. S.; Makarov, N.; Barlow, S.; Marder, S. R.; Perry, J. W., Nonlinear Optical Pulse Suppression via Ultrafast Photoinduced Electron Transfer in an Aggregated Perylene Diimide/Oligothiophene Molecular Triad. The Journal of Physical Chemistry A 2014, 118 (1), 110–121.
- Chen, L.; Zhang, K.; Tang, C.; Zheng, Q.; Xiao, Y., Controllable and Stepwise Synthesis of Soluble Ladder-Conjugated Bis(Perylene Imide) Fluorenebisimidazole as a Multifunctional Optoelectronic Material. The Journal of Organic Chemistry 2015, 80 (3), 1871–1877.
- Pagoaga, B.; Mongin, O.; Caselli, M.; Vanossi, D.; Momicchioli, F.; Blanchard-Desce, M.; Lemercier, G.; Hoffmann, N.; Ponterini, G., Optical and photophysical properties of anisole- and cyanobenzene-substituted perylene diimides. Physical Chemistry Chemical Physics 2016, 18 (6), 4924–4941.
- Cao, L.; Zhang, D.; Xu, L.; Fang, Z.; Jiang, X.-F.; Lu, F., Perylenediimide–Benzanthrone Dyad: Organic Chromophores with Enhanced Third-Order Nonlinear-Optical Activities. European Journal of Organic Chemistry 2017, 2017 (17), 2495–2500.
- Cao, L.; Xu, L.; Zhang, D.; Zhou, Y.; Zheng, Y.; Fu, Q.; Jiang, X.-F.; Lu, F., D-A dyad and D-A-D triad incorporating triphenylamine, benzanthrone and perylene diimide: Synthesis, electrochemical, linear and nonlinear optical properties. Chemical Physics Letters 2017, 682, 133–139.
- Cai, Z.; Vázquez, R. J.; Zhao, D.; Li, L.; Lo, W.-y.; Zhang, N.; Wu, Q.; Keller, B.; Eshun, A.; Abeyasinghe, N.; Banaszak-Holl, H.; Goodson, T.; Yu, L., Two Photon Absorption Study of Low-Bandgap, Fully Conjugated Perylene Diimide-Thienoacene-Perylene Diimide Ladder-Type Molecules. Chemistry of Materials 2017, 29 (16), 6726–6732.
- Garoni, E.; Nisic, F.; Colombo, A.; Fantacci, S.; Griffini, G.; Kamada, K.; Roberto, D.; Dragonetti, C., Perylenetetracarboxy-3,4:9,10-diimide derivatives with large two-photon absorption activity. New Journal of Chemistry 2019, 43 (4), 1885–1893.
- Madu, I. K.; Jiang, H.; Laventure, A.; Zimmerman, P. M.; Welch, G. C.; Goodson, T., Impact of Ring-Fusion on the Excited State Decay Pathways of N-Annulated Perylene Diimides. The Journal of Physical Chemistry C 2021, 125 (19), 10500–10515.
- Long, X.; Wu, J.; Yang, S.; Deng, Z.; Zheng, Y.; Zhang, W.; Jiang, X.-F.; Lu, F.; Li, M.-D.; Xu, L., Discovery of and insights into one-photon and two-photon excited ACQ-to-AIE conversion via positional isomerization. Journal of Materials Chemistry C 2021, 9 (35), 11679–11689.
- Feng, W.; Liu, K.; Zang, J.; Xu, J.; Peng, H.; Ding, L.; Liu, T.; Fang, Y., Resonance-Enhanced Two-Photon Absorption and Optical Power Limiting Properties of Three-Dimensional Perylene Bisimide Derivatives. The Journal of Physical Chemistry B 2021.
- Xu, B.; Zhang, J.; Fang, H.; Ma, S.; Chen, Q.; Sun, H.; Im, C.; Tian, W., Aggregation induced enhanced emission of conjugated dendrimers with a large intrinsic two-photon absorption cross-section. Polymer Chemistry 2014, 5 (2), 479–488.
There is NO Competing Interest.
- Supportinginformation.docx
supporting information
- floatimage6.jpeg
TOC graphic
{kind=link}