Polymer nanoparticles are the most promising vectors for targeted drug delivery due to their high biocompatibility, a wide spectrum of materials available for synthesis, and the ease of modification with molecules of different origins and functionality[30]. Among the wide range of natural and synthetic polymer materials for the design of therapeutic nanoparticles (such as protein-based polymers, polyphosphates, polyamides, polysaccharides, poly-lactic-co-glycolic acid) PLGA is the most popular polymer commonly used for biomedical and fundamental research application25. PLGA has been already approved by FDA for the therapeutic purposes and acts as a unique polymer for drug delivery [31, 32]. PLGA is a co-polymer of fully biocompatible and biodegradable lactic and glycolic acids and has already demonstrated remarkable results in clinical trials as an excellent candidate for drug delivery and treatment [33, 34].
To track the nanoparticles inside the organism, monitor their delivery and biodistribution, and use them for diagnostic tasks, we incorporated the fluorescent dye, namely, Nile Blue into the nanoparticle structure. Nile Blue is a fluorescent dye from the benzophenoxazine family with high fluorescence, high quantum yield, and excellent photostability [35]. The maximum excitation of the dye in dimethyl sulfoxide is 636 nm, and the maximum emission is 669 nm, thus entering the transparency window of biological tissues and making this dye promising for imaging applications in vivo [35]. Due to its lipophilic structure, Nile Blue was already used for several biological applications, e.g. for histology in vitro. Moreover, several in vivo studies have demonstrated the ability of this dye to accumulate in tumor cells after i.v. administration [27, 36]. Despite the above-mentioned advantages and low cost, Nile Blue was unjustly underestimated in biology with a limited number of studies related to its usage.
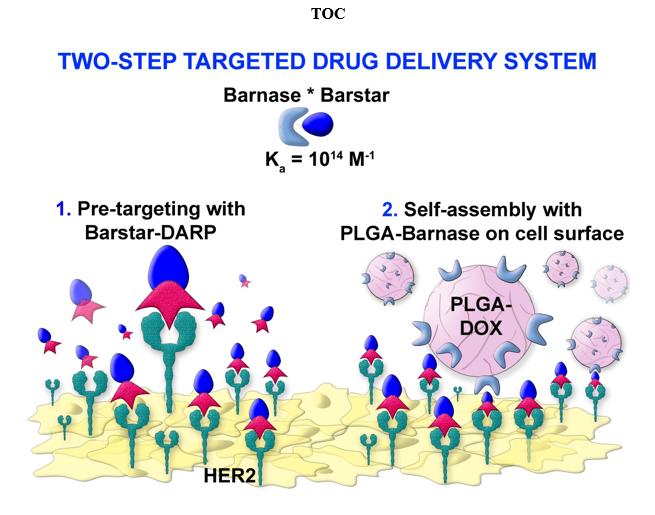
Here we describe the development of a two-step drug delivery system, based on polymer PLGA nanoparticles possessing both diagnostic and therapeutic properties. The delivery of these nanoparticles to HER2-overexpressing cancer cells is realized via proteinaceous Barnase*Barstar interface and HER2-recognizing scaffold protein DARPin9_29. The concept of targeted drug delivery suggests several advantages over standard chemotherapy, such as the decrease of the required dose of a drug, the improved drug penetration into the tumor, and the reduction of side effects [4]. However, the use of traditional targeting molecules for the therapeutics delivery, namely the use of monoclonal antibodies, often leads to a wide spectrum of side effects: i) considerable size of antibodies (150 kDa, 7-14 nm) often does not allow to modify the nanoparticle surface with the required number of IgG molecules for the efficient delivery to cells, ii) post-translational modifications of IgGs require biotechnological production in mammals which is time-consuming and expensive, iii) constant domains of the heavy chains have effector functions that may lead to phagocytosis without participating in the selective target recognizing, or can cause unwanted immunomodulation in vivo; iv) the presence of cysteines in the antibody molecule and glycosylation, which play an important structural role [14, 37, 38].
The artificial scaffold polypeptides were used in this study to target the nanoparticles toward cancer cells instead of the traditionally used monoclonal antibodies. In the last two decades, targeted polypeptide scaffold molecules of non-immunoglobulin nature obtained by phage, cellular or ribosomal display technologies, seem to be more effective tools for the delivery of nanoparticles and other substances to the target cells in the tumor site. The most promising synthetic scaffold proteins for the targeted delivery are DARPins (synthetic derivatives of cytoskeleton protein of drosophila – ankyrin) [16, 39–42], monobodies (derivatives of human fibronectin FN3 domain) [43], anticalins (derivatives of lipocalins) [44], avimers (derivatives of extracellular receptor A-domain) [38], affibodies (derivatives of highly stable domain B of staphylococcal protein A) [45] and others. Designed ankyrin repeat protein, or DARPin, was used in this study for the following reasons: it has a small size (14 kDa), high affinity to the molecular target, here receptor HER2 (KD = 3.8 nM), low immunogenicity, exceptional thermodynamic stability, and absence of cysteines in its structure [16, 46, 47]. Equally important is the ease of large-scale biotechnological production, in contrast to full-size antibodies [16, 48]. All these properties simplify genetic engineering and the creation of multispecific fusion proteins, which allow not only targeting different structures to the cells with certain molecular profiles but also realizing their own diagnostic and therapeutic functions [22, 25, 39, 42, 49]. Here we used DARPin9_29 genetically fused with Barstar which showed highly specific binding to receptor HER2 and allowed us to realize a two-stage delivery system based on Barnase*Barstar interface.
Table 1
The interfaces used for the drug delivery systems with pre-targeting step.
|
Delivery system
|
Immunogenicity
|
Sterical hindrance
|
Ka
|
Representation in mammals
|
|
Barnase*
Barstar
|
✓Both proteins are not immunogenic (unpublished data).
|
✓Proteins are comparable in size (12 and 10 kDa), and therefore steric hindrances should not arise[23].
|
1014 М–1 [50]
|
✓Both proteins were isolated from bacteria and are not represented in mammals [50].
|
|
Streptavidin*biotin
|
✗Streptavidin is highly immunogenic [51].
|
✗The significant difference in the size (56 kDa and 244 Da) of the molecules can cause steric hindrance: if biotin is bound to a non-smooth surface, then streptavidin will not be able to recognize it. This imposes restrictions on the use of this system in nanomedicine [52–54].
|
1015 М–1 [55]
|
✗Biotin, or vitamin H, is presented in the blood of mammals which may cause obstacles for appropriate interaction of streptavidin*biotin [56, 57].
|
|
Hapten*
antibody
|
✗Antibodies are immunogenic and may have effector functions which make them not the best candidate for long-term treatment [58].
|
✗IgG is 150 kDa protein while hapten is a low-molecular compound, so that steric difficulties may arise when the components interact [58, 59].
|
105-1010 М−1 [60]
|
✗Antibodies are presented in blood and have effector functions as critical participants in the immune defense [14].
|
|
Nucleic acids:
i) DNA*DNA
ii) RNA*RNA
iii) mirror-imaged oligonucleotides
iv) phosphorodiamidate morpholino oligomers
v) peptide nucleic acids
vi) locked nucleic acid
|
✓It was proposed that nucleotides as natural, presented in all organisms molecules with a high charge will be not immunogenic [61].
✓It was shown that some mirror-imaged oligonucleotides, as well as phosphorodiamidate morpholino oligomers and peptide nucleic acids are not immunogenic [62–65].
|
✗Nucleotides are small molecules (less than 500 Da). Since the entire sequence of nucleotides is vital in recognition, it can cause interaction problems if a non-smooth surface is used.
|
Depends on the base pair number
|
✗The presence of nucleases in the serum is an obstacle in the development of the system based on oligonucleotides due to the fast degradation [61].
✓Due to their artificial origin mirror-imaged oligonucleotides, phosphorodiamidate morpholino oligomers and peptide nucleic acids are resistant to the degradation by the nucleases, peptide and locked nucleic acids are also resistant to protease digestion [63, 65–68].
|
|
Click chemistry
|
✓Molecules used in bioorthogonal chemistry are supposed to be not highly immunogenic [69].
|
✗Molecules used in bioorthogonal chemistry are small which can cause interaction problems if a non-smooth surface is used. Also, this approach requires preliminary chemical modification of the delivered molecules, which proceed not completely steric and not with 100% yield.
|
✓✗Covalent bonding is stronger than affinity interaction but is not reversible.
|
✓Molecules used in bioorthogonal chemistry are not represented in mammals [66].
|
|
SpyTag/
SpyCatcher
|
N/A
|
✓Proteins are comparable in size (15 and 13 kDa), and therefore steric hindrances should not arise[70].
|
✓✗ Covalent bonding is stronger than affinity interaction but is not reversible.
|
✓Both proteins were isolated from bacteria and modified by bioengineering and are not represented in mammals [71].
|
Several two-stage DDSs are currently in use and under development (see Table 1). As shown in Table 1, Barnase*Barstar protein pair outperforms other two-stage systems which makes it a unique tool for the design of multifunctional biomedical products. Barstar (10 kDa) is a natural inhibitor of bacterial ribonuclease Barnase (12 kDa) [23]. These proteins have an extremely high binding affinity (association constant Kaff ~ 1014 М–1) and fast interaction kinetics (rate constant of complex formation kon ~ 108 М–1s–1), at the same time these proteins are not presented in mammals, which allows them to be used in blood flow without any interaction with endogenic components of blood [50, 72]. In the present study, Barstar-DARPin9_29 was used as the first component of the two-stage DDS and nanoparticles modified with Barnase served as the second component. It is also important to note, the lack of immunogenicity of all compounds used here – Barnase, Barstar, and Barstar-DARPin9_29.
Previously we showed the versatility of the Barnase*Barstar protein system for a wide range of applications including targeted delivery of both protein molecules, nanoparticles, and different supramolecular structures. In particular: i) the stability of the Barnase*Barstar protein complex under severe conditions (low pH, high temperature, and presence of chaotropic agents) was demonstrated that opens up more possibilities for using this system in any conditions both in vitro and in vivo [73]; ii) the successful labeling of HER2-overexpressing cancer cells in vitro with the self-assembled structures consisting of the magnetic particles and quantum dots using Barstar and scFv-Barnase-scFv construct (directed toward HER2 antigen) [21] and in vivo with radiolabeled 4D5 scFv–Barnase and 4D5 scFv–diBarnase [23] were shown; iii) a universal delivery system based on Barnase-Barstar and SiO2-binding peptide was developed [25]; iv) bispecific antibodies against HER1 and HER2 antigens using 425scFv-Barstar and 4D5scFv-Barnase [74] were obtained and utilized for imaging of cancer cells with overexpression of these receptors [75]. All the described studies confirm the versatility of the Barnase*Barstar interface for the wide range of biological applications that require the self-assembly of different structures in different conditions. In this work, using Barstar-DARPin9_29 and Barnase-conjugated polymer PLGA nanoparticles loaded with fluorescent dye Nile Blue and chemotherapeutic drug doxorubicin, we showed successful labeling and killing of HER2-overexpressing cells. The use of such two-step DDS allows decreasing the necessary dose of the doxorubicin needed to cause cancer cell death by one level of magnitude.
{kind=link}