Calcium imaging of ex vivo tendon fascicles during stretching
Tendon fascicles were gently extracted from the tail of skeletally mature 14-18 week old female Wistar rats (approval by the Veterinary Office of the Canton of Zurich, ZH235/16). Rat tail tendon fascicles were stained with 5 μM Fluo-4 AM (Thermo Fisher Scientific F14217) for 2 h at 29° C and 3% O2 in a modified Krebs-Henseleit solution (KHS) containing 126 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3 and 10 mM glucose. Single tendon fascicles were subsequently mounted on a custom-designed tensile stretching device (equipped with two linear motors and a 20 N load cell) that was placed on the stage of an iMic widefield microscope (Thermo Fisher Scientific) (56). During stretching protocols, fascicles were continually perfused with KHS that was preheated to 29° C and steadily aerated by a gas mixture containing 95% N2 and 5% CO2 to maintain 3% O2 and constant pH levels. Images were acquired with a 10x (N.A. 0.4) objective with excitation set at 488 nm wavelength and 100 ms exposure time. Prior to the stretching protocols, fascicles were preconditioned 5 times to 1.0% initial length L0 (10mm mounting length from clamp-to-clamp). The cross-sectional area was measured at crimp disappearance and L0 was defined at 1MPa tissue stress. Tissue strain was defined as (L-L0)/L0 in %, with sample length L. Tissue stress was calculated by dividing the force values through the cross-sectional area.
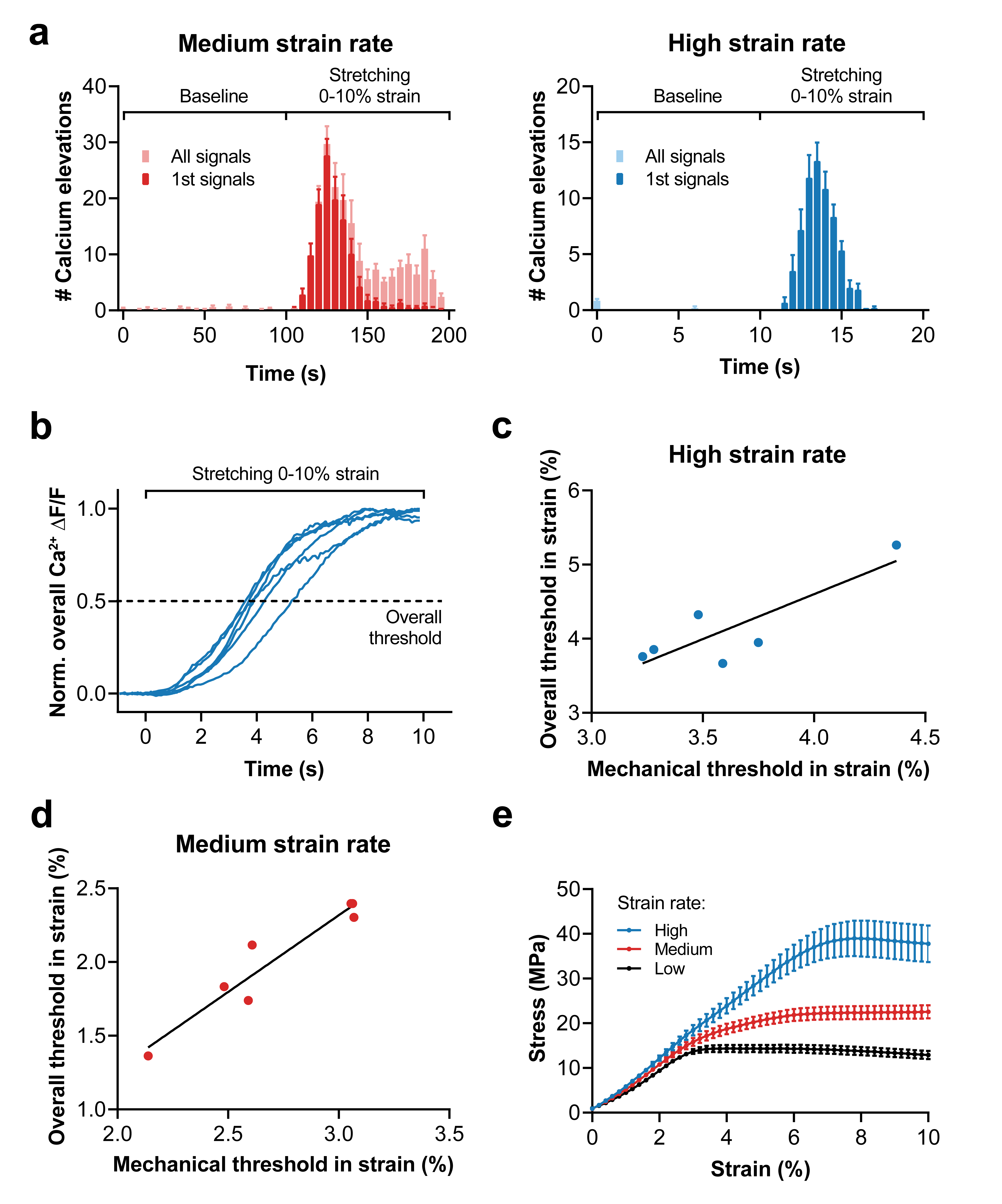
To investigate the cellular response to tissue stretching, single fascicles were stretched at three different strain rates (low 0.01% strain/s, medium 0.1% strain/s, high 1.0% strain/s) from 0 to 10% strain. Baseline activity was investigated at the preload of 1 MPa prior to initiating the stretching protocol. At medium and high strain rate, time lapses of one z-plane were recorded. At low strain rate, series of image stacks (50x2 µm) were acquired and subsequently deconvolved using Huygens Professional 18.10 (Scientific Volume Imaging) to quantify the micro-mechanical environment (at the cellular/collagen fiber level) needed to trigger intracellular calcium signals. To examine the time lag between the mechanical stimulus and the downstream calcium signals, tendon fascicles were subjected to one cycle of 2.7% strain at 30% strain/s.
Time lapse images were analyzed with the following steps. Cell movements were tracked with Imaris 7.7 (Bitplane AG, Switzerland) and the exported displacements were further used in a custom software (Matlab R2016a) that divided the images into 6x6 subimages and applied motion correction in each subimage. Calcium events were automatically detected and measured in the stabilized subimages using CHIPS (Cellular and Hemodynamic Image Processing Suite) (57). The interfiber sliding was calculated from the relative displacements (in axial direction) of the cells on adjacent fibers (e.g. fiber u and v): , n and m are the respective cell numbers of the adjacent fibers, L0 is the fiber length (58).
Fluorescence Lifetime Imaging (FLIM) of tendon fascicles
Isolated rat tail tendon fascicles were stained with 20 μM cell-permeable OGB-1 (Thermo Fisher Scientific O6806) for 2 h in KHS at 29° C and 3% O2 and mounted on our tensile stretching device placed on the stage of a two-photon microscope and continually perfused with KHS during imaging. FLIM was performed on an upright Leica TCS SP8 FLIM two-photon microscope equipped with a tunable (680-1300 nm) 80 MHz infrared laser (Insight DS+ Dual from Spectra Physics) and four non-descanned FLIM enabled hybrid detectors. On the non-descanned detectors the following emission band pass filters were used: 460/50 nm, 525/50 nm, 585/40 nm, 650/50 nm. Data acquired from the channels 525/50 nm and 585/40 nm were used for the FLIM analysis. A 2.6 mm working distance Leica HC IRAPO 25x/1.0 water immersion objective was used for imaging. Time-correlated single-photon counting (TCSPC) was performed using a six channel Picoquant HydraHarp 400 together with the Picoquant Symphotime 64 software package. Femtosecond infrared laser pulses allowed for efficient two-photon fluorescence excitation and emission from a thin focal plane in the core of a tendon fascicle (ca. 30-100 μm deep in the tissue). A laser wavelength of 915 nm and a maximum laser power of 12.5 mW measured after the objective were used to avoid any signs of phototoxicity. To avoid photo-induced effects as well as statistical pile-up effects in the TCSPC histograms, the photon count rates on the detectors were always kept below 1% of the excitation rate. For fast sequential imaging an image size of 512 px in width and 80-110 px in height was used with a scanner frequency of 400 Hz.
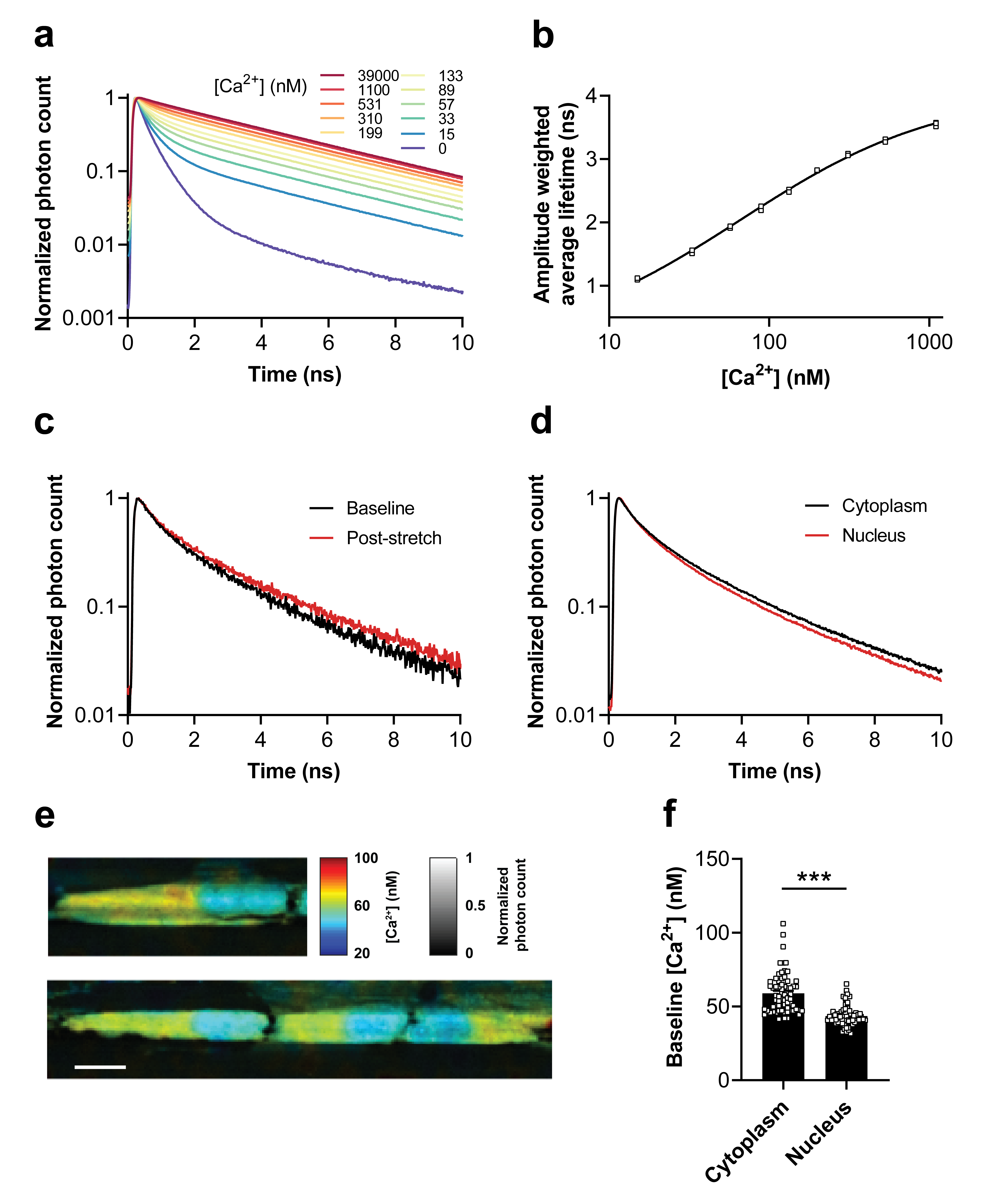
The calcium calibration buffer kit from Thermo Fisher Scientific (C3008MP) was used in combination with the cell impermeable Ca2+ indicator OGB-1 at 1 μM to calibrate the [Ca2+] readout (Fig. S2a). Temperature and pH were measured and considered by finely adjusting the estimated [Ca2+] using Chris Patton’s WEBMAXC program (http://web.stanford.edu/~cpatton/webmaxcS.htm) (32). The TCSPC histograms were fitted using a double-exponential tailfit within a time gate of 10ns using the Picoquant Symphotime software. From the tailfit, we calculated the amplitude weighted average lifetime, which was used throughout the study as a readout for [Ca2+] using a suitable calibration function acquired from fitting a nonlinear Hill function to the OGB-1 calibration data (Fig. S2b).
To investigate baseline [Ca2+] in tenocytes, tendon fascicles were imaged at the preload of 1 MPa after a preconditioning. Average image acquisition times were 120 s. A cellular compartment dependent region of interest analysis was performed to determine calcium concentrations in the cytoplasm and in the nucleus. The detected photons within the selected compartments were aggregated to obtain an overall [Ca2+] estimate for that region. Ca2+ concentrations during calcium signals were examined with two different approaches. First, time-lapses were recorded after a stretch of the tendon fascicle to analyze the [Ca2+] during spontaneous calcium signals. Second, time-lapses were acquired pre- and post-stretch to study the increase in [Ca2+] induced by mechanical loading. In both cases, the stretching protocol consisted of a single cycle to 2.7% strain at 1.0% strain/s starting and ending at the preload. The time-lapses were taken over a period of 15 min. To monitor the calcium signals, we maximized the temporal FLIM resolution while keeping an appropriate spatial resolution and the total number of collected photons. Therefore, we adjusted the acquisition rate to 0.066 Hz, which was sufficient to estimate the [Ca2+] during calcium events because of their average duration of around 27 s. The photons were aggregated within the selected single cell area. Subsequent data analysis calculated the [Ca2+] during calcium events. For illustration purposes, we applied a 2x2 px binning and a Gaussian filter (σ = 3) to the pixel maps of the [Ca2+]-landscape. Data analysis and statistics, however, were performed using the raw data, not the filtered pixel maps.
Mathematical model for shear stress prediction in tenocytes
To predict the shear stress experienced by tenocytes during tissue stretching, we applied a numerical model that assumes cell heights (h) between 1-10 μm and that assumes individual tenocytes span the distance between two adjacent collagen fibers (59). Shear stress in tenocytes is generated by unilateral collagen fiber displacement, which leads to transverse displacement of the cell body. By definition, the shear stress (τ) arises through the application of a force (F) parallel to the cross-section over a certain surface (A) and is equal to the shear modulus (G) of the material multiplied by the shear strain (γ). The shear strain is defined as the transverse displacement (Δx) of the material divided by the initial height of the material (h) (Fig. 1i): . The shear modulus of a eukaryotic cell was previously estimated to be around G = 1.5 Pa (60) and the transverse material displacement (Δx) was calculated from the interfiber sliding (s) and the fiber length (lfiber = 900 μm). This enables the estimation of shear stresses acting on tenocytes resulting from unilateral collagen fiber sliding.
Primary human and rat tenocyte cultures
Tendon cells were isolated either from fragments of human tendons (flexor hallucis longus, gracilis and semitendinosus) collected from female and male patients (between 24 and 58 years of age) undergoing treatment at the University Hospital Balgrist (permission 2015-0089 from the institutional review board of the Canton of Zurich and patient-informed consent) or from tail tendon fascicles of skeletally mature 14-18 week old female Wistar rats (approval by the Veterinary Office of the Canton of Zurich, ZH235/16). Next, tendon tissues were digested with 2mg/ml collagenase-D (Roche 11088866001) in Dulbecco’s modified Eagle’s medium (DMEM/F12 D8437) supplemented with 1% Amphotericin B (Gibco 15290-018) and 1% Penicillin-Streptomycin (Sigma-Aldrich P0781) for ca. 6 h at 37° C in a humidified atmosphere of 5% CO2. Isolated cells were cultured on tissue culture plastic in DMEM/F12 with 10% heat inactivated fetal bovine serum (FBS, Gibco 10500) for 1-2 weeks and subsequently cryopreserved in liquid nitrogen until the start of the experiments.
Calcium imaging during application of shear stress on isolated tenocytes using flow chambers
Custom-made flow chambers were fabricated with the following procedure. A microscope slide was plasma treated, and 3 μl of polydimethylsiloxane (PDMS, Sylgard 184 Silicone Elastomer Kit, Dow Europe) was deposited in its center. Then a silanized PDMS stamp that was molded from the negative of the microgroove pattern (10 μm depth/ridge width/pitch) was placed on top. The assembly was subsequently cured at 70° C for 6 hours before detaching the stamps. PDMS microgrooves were chemically activated using two consecutive treatments of 0.1 mM N-sulfosuccinimidyl-6-(4′-azido-2′-nitrophenylamino) hexanoate (CovaChem 13414) in 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Seraglob K 2101) under ultraviolet light for 10 min each. Substrates were then washed three times with sterile phosphate buffered saline (PBS) and coated with 50 μg/ml collagen-I (Corning 354249) in PBS overnight at 4°C, before being washed three times with deionized water and air dried (61). Finally, a block of PDMS containing a 0.4 mm high, 5 mm wide and 30 mm long canal was glued on top of the microscope slide, centering the microgrooved patch in the middle of the canal. A fine layer of PDMS containing additional 0.1% platinum-divinyltetramethyldisiloxane (abcr 146697) was used as glue and cured at 45° C for two hours.
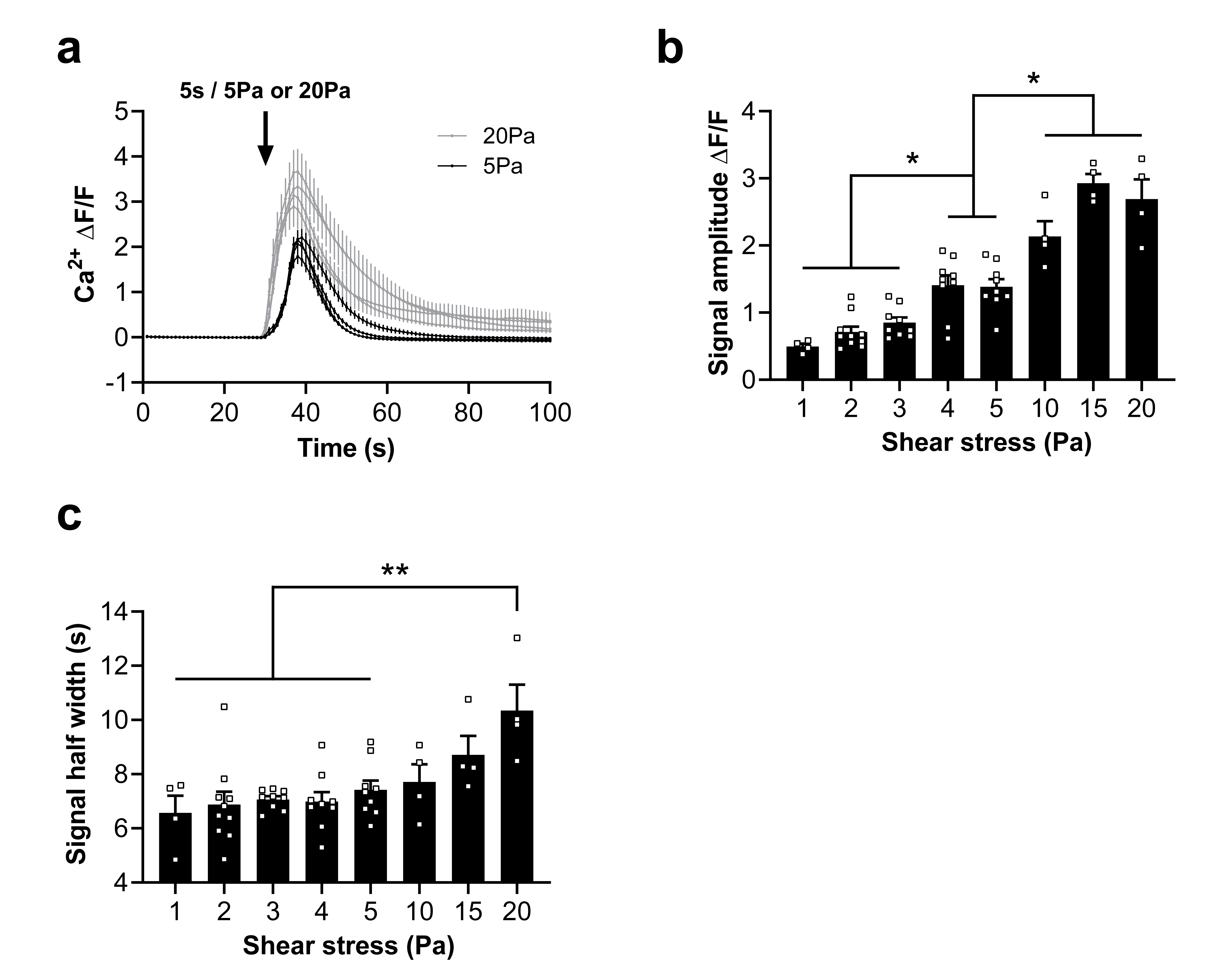
Tenocytes were seeded in the flow chamber at a density of 38’000 cells/cm2 and incubated at 29° C / 5% CO2 / 3% O2 overnight. Staining for 2 h with 1 µM Fluo 4-AM diluted in KHS containing 0.02% pluronic F-127 was performed before placing the flow chamber on the microscope stage and connecting it to a syringe pump (Cetoni, low-pressure module). Next, the flow chambers were flushed for a few minutes at a flow rate of 0.1 ml/min (resulting in negligible shear stress of ca. 0.01 Pa) with KHC that was preheated to 29° C and degassed to 3% O2 using a gas mixer. Appropriate flow rates resulting in specific shear stresses on the cell substrate were calculated using established formulas of fluid flow in rectangular channels (62). During shear stress experiments, image stacks (5x3 μm) were acquired with the iMic widefield microscope (10x objective) at a frequency of 1 Hz, a wavelength of 488 nm, and an exposure time of 100 ms.
Image analysis was done with an initial average intensity Z-projection of the image stacks, followed by a segmentation of individual cell bodies performed with a custom ImageJ script based on the fluorescence at the baseline, i.e. 30 s interval before application of the shear stress stimulus. The mean fluorescence intensity of each segmented cell was normalized to the average intensity measured at the baseline. A calcium signal in a cell was defined as such when the normalized fluorescence intensity (ΔF/F0) exceeded the baseline fluorescence intensity by 10 times the standard deviation of the baseline during a 20 s interval following shear stress exposure.
Generation of CRISPR/Cas9-mediated knockdown cells
Single guide RNAs (sgRNAs) against multiple candidate genes were designed with the CRISPRdirect online tool http://crispr.dbcls.jp (63). Only highly specific target sites were selected, the respective sequences are listed in Table S1. A non-targeting control sgRNA was chosen from the study of Morgens et al. (64) and checked for low targeting potential by BLASTN 2.8.0 search. Target sequences oligos were synthesized with BsmBI restriction site overhangs by Microsynth (Balgach, Switzerland) and then annealed and cloned into the lentiCRISPRv2 transfer plasmid, a gift from Feng Zhang (Addgene plasmid #52961; (65)), following the provided protocol of the Feng Zhang Lab.
Lentiviral particles were produced by co-transfection of the lentiCRISPRv2 plasmid, containing the respective gRNA-sequence, with the packaging plasmids pCMV-VSV-G (a gift from Bob Weinberg; Addgene plasmid #8454; (66)) and psPAX2 (a gift from Didier Trono; Addgene plasmid #12260) into HEK293T cells using Lipofectamine 3000 (Thermo Fisher Scientific L3000008) and following the manufacturer’s instructions.
For transduction, human and rat tenocytes were incubated for 24 h with supernatant containing the viral particles and supplemented with 8 µg/ml Polybrene. Subsequently, human and rat cells were selected with 3 µg/ml Puromycin (Gibco A1113803) for 3 days or with 4 µg/ml for 7 days, respectively. The efficiency of the knockouts was tested with quantitative real-time PCR, immunofluorescence and western blotting.
RNA isolation from tissues and cells and quantitative real-time PCR
Freshly isolated tissues were snap frozen in liquid nitrogen and subsequently homogenized with QIAzol lysis reagent (Qiagen 79306) using a cryogenic grinder (SPEXSamplePrep FreezerMill 6870). 1-bromo-3-chloropropane (Sigma-Aldrich B9673) was added to the tissue lysates at a 1:4 ratio, and the RNA containing aqueous phase was obtained using Phase Lock Gel - Heavy (LabForce 2302830). In vitro tenocytes were lysed with RLT/bME buffer. Subsequently, RNA from tissue and cell lysates was extracted using the RNeasy micro Kit (Qiagen 74004) following the protocol provided by the manufacturer. Quality and quantity of the RNA was measured with a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific).
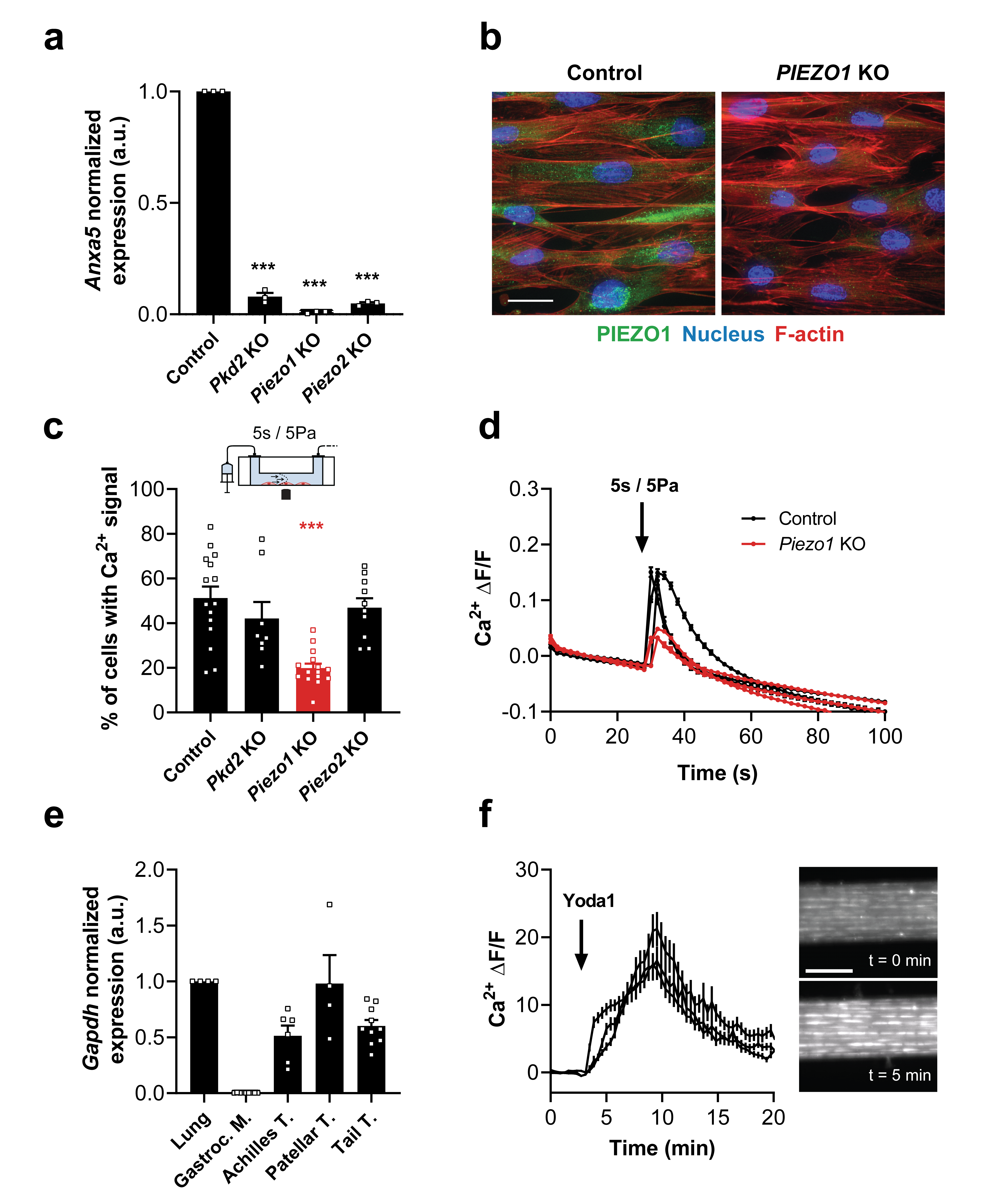
cDNA was synthesized from 500 ng of total RNA using a High-Capacity cDNA Reverse Transcription Kit with RNAse Inhibitor according to the manufacturer instructions (Applied Biosystems 4374966). Gene expression analysis was performed by quantitative real-time PCR with cDNA corresponding to 10 ng of starting RNA using the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific A25742). The samples were amplified using a StepOnePlus Real-Time PCR System (Applied Biosystems) with the following conditions: 95° C for 10 min followed by 40 PCR cycles of 95° for 15 s and 60° C for 1 min. All experiments were run with technical triplicates. Relative gene expression levels were quantified using the 2–ddCT method with either ANXA5 or GAPDH as reference gene. All primers are listed in Table S2.
Immunofluorescence
Cells seeded in flow chambers were fixed with 4% formaldehyde in PBS (Carl Roth 3105.2) for 20 min at room temperature and subsequently permeabilized with 0.1% Triton-X (Axonlab 10029070) and 0.5% bovine albumin serum (BSA, VWR P6154) in PBS for 10 min. Samples were incubated with specific primary antibodies in PBS with 3% BSA for 1 h and afterwards with a secondary fluorescently-labeled antibody. Between every step samples were washed three times with PBS. Primary antibodies were used against PIEZO1 (Novus Biologicals NBP1-78446 for human cells, 1:25; Alomone Labs APC-087 for rat cells, 1:300). Actin filaments were stained with Alexa Fluor 568 phalloidin (Thermo Fisher Scientific A12380, 1:200) and cell nuclei with NucBlue (Thermo Fisher Scientific R37605). Alexa Fluor 488 conjugated donkey anti-rabbit (Thermo Fisher Scientific A-21206, 1:100) was used as a secondary antibody. Immunofluorescence images were acquired with the iMic spinning disk confocal microscope using an oil-immersion 60x (N.A. 1.35) objective.
Western blotting
Cells were washed with PBS and lysed directly in the cell culture dish with 80 μl of 1x reduced Laemmli buffer (Fisher Scientific 15493939) and boiled for 5 min at 95°C. 15 μl of each sample was loaded onto a 4-15% Mini-PROTEAN TGX stain-free protein gel (Bio-Rad 4568086). Total protein was analyzed using the Criterion Stain-free imaging system (Bio-Rad) and subsequently transferred on polyvinylidene difluoride membranes using the Trans-Blot-Turbo Transfer System (Bio-Rad). Membrane blocking was carried out with 5% nonfat dry milk/TBS-T for 1 h at room temperature. The primary antibodies targeting PIEZO1 (Thermo Fisher Scientific MA5-32876, 1:500) and β-tubulin (MERCK Millipore MAB3408, 1:10’000) were diluted in 5% BSA/TBS-T and incubated overnight at 4° C. Next, the membranes were washed 3 times in TBS-T and incubated with the secondary antibody (anti-mouse, Sigma-Aldrich SAB3701073, 1:20’000) for 1 h at room temperature. Images were taken using UltraScence Pico Ultra Western Substrate (GeneDireX CCH345-B) and the ChemiDoc MP imaging system (Bio-Rad).
Tendon explants cultured in bioreactor and subjected to sham or Yoda1 stimulation
Rat tail tendon fascicles were freshly isolated and placed into culture medium (high glucose Dulbecco’s Modified Eagle’s Medium, Sigma Aldrich D6429, supplemented with 1% Penn/Strep, 200 μM ascorbic acid, Wako Chemicals 013-19641, and 1% N-2 supplement, Thermo Fisher Scientific 7001585). Each fascicle was cut in half, one half was used for mechanical testing at day 0, while the other half was cultured in our custom-made bioreactor at the preload (crimp disappearance, i.e. minimal mechanical load) and mechanically tested at day 16 (39). Distal and proximal samples were randomly distributed between the two days. Diameters were measured at day 0 and day 16 using a 10x objective (Motic AE2000). Cultured fascicles underwent either sham or 5 μM Yoda1 stimulations for 30 min on days 0, 3, 6, 9 and 12 post-isolation. Following the 30 min treatment, fascicles were washed once with medium, then resuspended in medium and incubated at 29° C, 5% CO2 and 3% O2. Ramp-to-failure experiments were performed to assess the biomechanical properties. Samples were preloaded to 0.04 N and preconditioned 5 times to 1% strain. Subsequently, a ramp-to-failure was carried out at 1% strain/s. Fascicle stiffness was calculated in the linear region of the force-strain curves and fascicle strength was determined from the maximal force.
Biomechanical testing and analysis
The biomechanical properties of plantaris tendons from wild-type and Piezo1GOF mice (31) were investigated with ramp-to-failure experiments. Age-matched littermates (20-21 weeks old) were euthanized and stored at -80° C until the day of experiment (approval by the Institutional Animal Care and Use Committees of Scripps Research in accordance with the guidelines established by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)). After thawing, plantaris tendons were carefully isolated and tested in uniaxial tension using a custom clamping technique on a universal testing machine that recorded force-displacement data (Zwick Z010 TN, 20 N load-cell) (67). During testing, tendons were kept in a custom chamber filled with KHS, preloaded to 0.1 N (initial length L0 corresponding to 0% strain) and preconditioned 5 times to 1% strain (preload reapplied after every cycle). Subsequently, samples were ramped to failure at a constant strain-rate of 1% strain/s. The diameter was measured in microscopic images of the plantaris tendons (4x objective, Motic AE2000). Tendon stiffness was calculated in the linear region of the force-strain curves and tendon strength was determined from the maximal force.
Transmission electron microscopy
Plantaris tendons were freshly isolated from age-matched littermates (29-34 weeks old) euthanized in the middle of the day (normal light/dark cycle) and sequentially fixed with 2.5% glutaraldehyde (Sigma-Aldrich G5882) in 0.1 M sodium cacodylate buffer (pH 7.2), with 1% OsO4 in 0.1 M sodium cacodylate buffer at room temperature and with 1% uranyl acetate in H2O at room temperature for at least 1 hour per step. Samples were rinsed 3 times between the fixation steps and finally with H2O prior to dehydration in an ethanol series and embedding in Epon. Ultrathin (70 nm) sections were post-stained with Reynolds lead citrate and imaged in a FEI Talos 120 at 120 kV using a bottom mounted Ceta camera (CMOS, 4k x 4k pixels) using MAPS software (Thermo Fisher Scientific). Segmentation of the cross-sectional area of collagen fibrils was performed with the Trainable Weka Segmentation Fiji plugin (68).
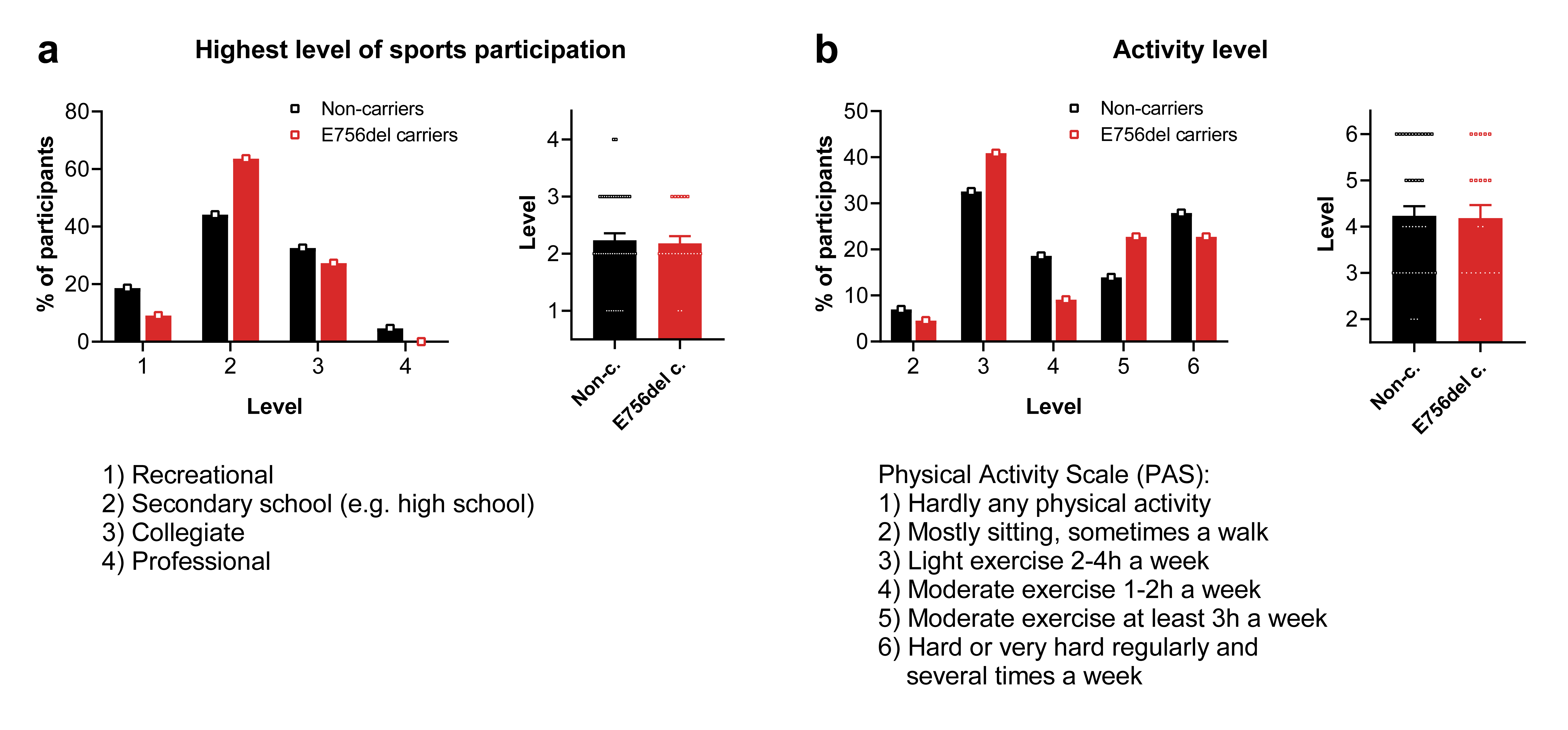
Participants of the human study - activity level and sports participation
Healthy self-reported African American participants (at least 18 years old) were enrolled after approval by the Institutional Review Board of the University of Delaware (ID-1420251-3) and written informed consent. A clinical evaluation with ultrasound imaging was carried out to ensure that the subjects had no underlying pathologies in their Achilles tendons. Additionally, the Victorian Institute for Sports Assessment - Achilles questionnaire (VISA-A) was applied to confirm that the Achilles tendons were healthy (69). Participants reported their highest level of sports participation (recreational, secondary school, collegiate or professional) and their history of sports participation. The Physical Activity Scale (PAS) was used to assess the subject’s reported current physical activity level (70).
Ultrasound-based assessment of the human Achilles tendon morphology
B-mode ultrasound imaging (LOGIQ e ultrasound system (GE Healthcare, USA) with wide‐band linear array probe (5.0‐13.0 MHz)) was used to analyze the Achilles tendon morphology Briefly, Achilles tendon thickness and cross‐sectional area were measured at a distance of 2.5 cm from the calcaneal osteotendinous junction (axial direction) in the images acquired parallel and perpendicular to the fiber orientation (each 3 repetitions), respectively (71). Achilles tendon length was measured as the distance from the calcaneal osteotendinous junction to the myotendinous junction of the gastrocnemius and soleus muscles using extended field of view (71). The software OsiriX MD (Pixmeo SARL, Switzerland) was used to analyze the ultrasound images.
Functional performance tests
To investigate the jump heights, we used the MuscleLabÔ (Ergotest Innovation, Norway) light mat measurement system, which creates an infrared light field 4 mm above the floor and records beam interruptions. Participant height and weight were recorded prior to jumping tests. Jump height was calculated by MuscleLabÔ using participant weight, ground contact time, and flight time. Participants performed two different jump tests. In each test the participants were asked to place their hands behind their back and to jump as high as possible. The first test was a single leg countermovement jump (CMJ) in which the participants started by standing on the floor on one leg then quickly bent the knee before jumping straight up as high as possible (72). This was repeated three times with each leg. The second test was a single leg drop countermovement jump (DCMJ), in which participants jumped off of a 20 cm high box and then jumped vertically as high as possible (72). For each leg, the average height of three trials was used for analysis.
Genotyping of the human PIEZO1GOF (E756del) mutation
Saliva samples were collected using Oragene DNA collection kits (OG-500, DNA Genotek). Genomic DNA isolation was performed according to the manufacturer instructions using the prepIT-L2P reagent (PT-L2P, DNA Genotek) included in the kit. The region containing the E756 locus was amplified with PCR (forward primer 5’ CAGGCAGGATGCAGTGAGTG 3’ and reverse primer 5’ GGACATGGCACAGCAGACTG 3’) (31). This amplicon (ca. 200 bp) was sequenced using both primers to identify non-carriers and E756del carriers.
Statistical analysis
For multiple-comparisons, data were analyzed with one-way ANOVA (Tukey’s or Dunnett’s test). Inter-group comparisons were performed with two-tailed Student’s t test or two-tailed Mann-Whitney test. For mouse and human data, n = number of animals or participants, at least n = 4 was used. Analyses using linear mixed effects models (lme4 package in R) were conducted for the biomechanical experiments with mouse tendons (mouse ID as random effect and litter as fixed effect, Bonferroni-Holm correction) and for the human jumping data (subject ID as random effect and leg as fixed effect). Age, height, weight, highest level of sports participation and activity level of the participants were tested as covariates. Significance levels were set as *p < 0.05, **p < 0.01, ***p < 0.001. In the figures, data are represented as means ± SEM. Analyses performed with GraphPad Prism 8.2 or RStudio v1.1.383.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}