Experimental animals and tissues
Six LT sows and six LR sows were artificially inseminated with semen from the same breed boars, respectively. All sows were slaughtered at 35 dpc after insemination, and embryos were collected as previously described. For each embryo, the longissimus dorsi muscle (LDM) tissues were isolated, then, digested to isolate embryonic muscle progenitors or fixed to prepare paraffin sections of tissue, or frozen in liquid nitrogen for further use.
Embryonic muscle progenitors isolation and culture condition
Embryonic muscle progenitors were isolated from the LDM of embryos of two breeds at 35 dpc. The isolated LDM was cleaned free of connective tissues, minced and digested with 0.2% Collagenase type I (Sigma, Shanghai, China) solution at 37°C water bath for 2h to get sufficient cells. Mixed cells was pre-plated 2 h in 5.0 ml growth media on a culture dish to remove fibroblasts and then transferred to a new culture dish for attachment. Cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 20% (v/v) fetal bovine serum (FBS), 1% penicillin–streptomycin antibiotics and 0.5% chicken essential extract (growth medium, GM). For experiments, muscle progenitors were sub-cultured onto 12-well plates at densities of 1.0 × 104 cells per well. Cells were switched into DMEM with 2% horse serum (differentiation medium, DM) after reaching 100% confluence to induce differentiation.
C2C12 cells were purchased from American Tissue Culture Collection (CATCC), cultured in DMEM with 10% FBS, and 1% penicillin-streptomycin (growth medium, GM) at subconfluent density. To induce differentiation, cells were switched into DM after reaching 100% confluence. All cells were cultured at 37 °C in a humidified atmosphere of 5% CO2.
In vitro Notch activation or inhibition
For Notch activation, C2C12 cells were treated with the Jagged1 peptide (CDDYYYGFGCNKFCRPR) or a control scrambled peptide (RCGPDCFDNYGRYKYCF) (GenScript USA, Inc.) (20 µg/ml) for 48 or 72 h in proliferation or myogenic differentiation assay [24, 25]. For Notch inhibition, embryonic muscle progenitors cultured in GM were treated with DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester) (20 µM, solved in DMSO) for 60, 72, 84, 96, 108, 120 or 132 h according to different assays; embryonic muscle progenitors cultured in DM were treated with DAPT for 144 h; control cells were treated with carrier (DMSO) only [26].
siRNAs, plasmids and transfection
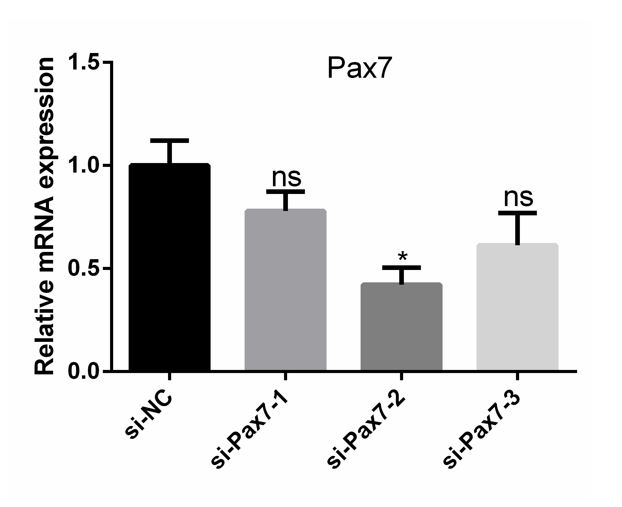
For RNA interference, negative control siRNAs (siNC) and three stealth mouse Pax7 siRNAs were purchased from Invitrogen (Thermo Fisher Scientific, USA) (Table. S1). siPax7-2 was the most efficient (Fig. S1). So, it was used in all of the following analysis. For Pax7 expression vector, mouse Pax7 CDS sequence was inserted into pcDNA3.1 vector (Invitrogen, Shanghai, China). C2C12 cells were transfected with siRNAs or plasmids using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instruction. All transfections were performed in triplicate for each experiment.
Explant
Forelimbs from LR 35 dpc embryos were cultured in 12-well plates in BGJb medium (Life Technologies), without FBS, with 200 µg/ml ascorbic acid (Life Technologies) and 1% penicillin–streptomycin antibiotics. For Notch inhibition, forelimbs originating from the same embryo were immediately treated with 20 µM DAPT or DMSO carrier for 30 h. Then, treated and control forelimbs were cleaned and collected for further analysis.
Western blot
Protein extracts were obtained from incubating cultured cells, LDM or forelimbs homogenates in lysis buffer (150 mmol/L NaCl, 50 mmol/L Tris, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, pH 8.0) supplemented with protease inhibitor phenylmethanesulfonyl fluoride (PMSF, Thermo Scientific) on ice until protein was released completely. Total extracts were separated by SDS-PAGE on 10% (w/v) polyacrylamide gels, transferred onto 0.45 μm PVDF membrane (Roche). After being blocked in 4% bovine serum albumin (BSA) for 1-2 h, the membranes were incubated with specific primary antibodies at 4 °C overnight, then incubated with secondary antibodies for 1 h at room temperature. Blots were visualized using an enhanced chemiluminescence (ECL) detection kit (FDbio, Hangzhou, China). β-Tubulin and GAPDH were used as internal controls. Antibodies are listed in Table S2.
RNA extraction and real‐time quantitative PCR
Total RNA was extracted from cultured cells, LDM or forelimbs according to the instructions of TRIzol® Reagent (Invitrogen, Shanghai, China), then cDNA was synthesized from 1 μg total RNA using a reverse-transcription Kit (Promega, Beijing, China). The real-time quantitative PCR (qPCR) was performed using a SYBR Green qPCR Kit (Genestar, Beijing, China), detected on the LightCycler 480 II system (Roche, Basel, Switzerland). The primers used for qPCR were given in Table S3. Gapdh was used as internal control and all reactions were run in triplicate.
Immunofluorescence
Cultured cells were fixed in 4% paraformaldehyde for 10 minutes, permeabilized in 0.5% Triton X‐100 for 15‐20 minutes. After being blocked with 4% BSA in Tris‐buffered saline with Tween (TBST) for 1 hour, the cells were incubated with primary antibodies overnight at 4°C, followed by incubation with secondary antibodies for 1 hour at room temperature. The nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI; 1:1000 in PBS). Antibodies are listed in Table S2. Immunostaining images were obtained via fluorescent reverse microscopy (Nikon, Tokyo, Japan).
Immunohistochemistry
LDM or forelimbs were fixed in 4% paraformaldehyde for 19 h at 4°C, then dehydrated using gradient alcohol and embedded with paraffin. Paraffin embedded samples were cut into 5-μm sections. The paraffin sections were placed in an oven at 64°C for 30 minutes and immediately moved to xylene for dewaxing. Rehydrated in gradient alcohol, antigen repaired using citrate antigen retrieval solution. Finally, immunofluorescence was performed using IHC Kit (Abcam, Cambridge, England) according to the manufacturer’s instruction. Immunostaining images were obtained via fluorescent reverse microscopy (Nikon).
Statistical test
Immunostainings were performed on at least three embryos of each group. Images of immunostainings were randomly selected for analysis. The fusion index was calculated as the percentage of nuclei in fused myotubes out of the total nuclei. Data are presented as mean±sem or mean from at least three independent experiments. Statistical differences between groups were tested using an unpaired two-tailed Student’s t-test. Values of P<0.05 was considered as significance.
{kind=link}
{kind=link}