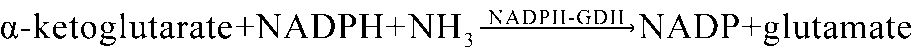3.1. Temperature and humidity index
As shown in Fig. 1, there were three times daily changes in THI values in the morning (08:00), noon (14:00), and evening (20:00) during the experimental period. The average daily THI values during the experimental period were higher than 79 for all 22 experimental days.
3.2.Body temperature, respiratory rate, rumen fermentation, and nutrient digestibility
Dietary supplementation with CrPyr decreased the body temperature of beef cattle (Fig. 2a, P < 0.05), but did not affect the respiratory rate of beef cattle (Fig. 2b). Diet supplemented with CrPyr increased the ruminal pH value (Fig. 2c, P < 0.05) and MCP concentration (Fig. 2d, P < 0.05). No effect was observed on the contents of NH3-N (Fig. 2e), total VFA (Fig. 2f), or individual VFA (Fig. 2g). As shown in Fig. 2h, CrPyr supplementation increased the crude fat digestibility (P < 0.05). No difference was found between groups for the digestibility of the dry matter, organic matter, crude protein, neutral detergent fiber, and acid detergent fiber.
3.3. Diversity of rumen fluid microbiota of beef cattle as revealed by 16S rDNA high-throughput sequencing
The high-throughput sequencing technology was used to investigate the effects of CrPyr on microbial communities in the rumen of beef cattle. The sequences were clustered into 1306 OTUs with a similarity of 97%. Among these, a total of 1098 OTUs were common, 109 OTUs were specific for the EG, and the CG had 99 unique OTUs (Additional file 2: Fig. S1). The rumen microbiota α-diversity of the two groups was evaluated by the Ace index, Chao1 index, Shannon index, and Simpson index (Additional file 3: Table S2). According to the result, there was no significant differences in the α-diversity index between the two groups.
3.4. Composition of rumen fluid microbiota of beef cattle as revealed by 16S rDNA sequencing and metaproteomics
16S rDNA sequencing and metaproteomics were combined to investigate the effects of CrPyr on both the structure and function of rumen fluid microbiota of beef cattle. 16S rDNA sequencing revealed that bacteria belonging to the phyla Bacteroidetes and Firmicutes comprised most (the average coverage was ~95.4%) of the total bacteria in the rumen fluid microbiota of the CG and EG. The remaining bacteria were mainly members of Actinobacteria, Spirochaetes, and Verrucomicrobiota (Fig. 3a). The abundance of Verrucomicrobiota was significantly different between the two groups (P < 0.05). At the genus level, the Rikenellaceae_RC9_gut_group was the dominant bacteria, and the other abundant genera were Prevotella, NK4A214_group, Christensenellaceae_R-7_group, Prevotellaceae_UCG-003, and Succiniclasticum (Fig. 3b).
Label-free quantification (LFQ) proteomics was applied to investigate the rumen fluid microbiota proteins of beef cattle. A total of 716,150 MS/MS spectra were generated from the six rumen fluid samples. Of these, 11,361 peptides (16.2%) could be identified and assigned to 3579 proteins (Additional file 4: Table S3). They included 2997 proteins that were quantified in the rumen fluid microbiota of EG and CG. The number of overlapping proteins between the two groups was 2153 (71.8%), while 373 and 206 community-specific proteins were unique to the rumen fluid microbiota in EG and CG, respectively, the number of quantified proteins without quantitative value (refers to a protein expressed in samples with less than 2/3 in both groups) was 265. Furthermore, in the 2153 overlapping proteins, 121 differentially expressed proteins (DEPs) were identified in the EG compared with the CG, of which 67 proteins were up-regulated and 54 proteins were down-regulated (Fig. 4b, P < 0.05). Of the proteins identified, most were related to cell metabolism (3407). Others were related to organismal systems (188), human diseases (324), genetic information processing (612), environmental information processing (295), and cellular processes (125) (Fig. 4a).
The 16S rDNA sequencing and proteomic data revealed differences in the compositions of the dominant phyla. 16S rDNA sequencing revealed that the 16S rDNA relative abundances (abbreviated as 16SDA hereafter) of members of Bacteroidetes increased from 54.08 with CG to 56.89% with EG, the Firmicutes 16SDA decreased from 41.47 to 38.35% (Fig. 3a). Metaproteomics showed that the protein relative abundances (abbreviated as PRA hereafter) of members of Bacteroidetes in total quantified proteins decreased from 74.15 with CG to 66.30% with EG, the Firmicutes PRA increased from 25.85 to 33.70% (Fig. 3c). At the genus level, the proteomic data showed that members of Prevotella were the dominant proteins, and the other abundant proteins were members of Bacteroides, Clostridium, Ruminococcus, and Alistipes (Fig. 3d).
3.5. Composition and functional classification of differentially expressed proteins as revealed by metaproteomics
The metaproteomics data showed that all 700 (373 unique in EG + 206 unique in CG + 121 differential overlapping, Additional file 5: Table S4). DEPs were members of Bacteroidetes and Firmicutes (Fig. 3e). The Bacteroidetes PRA was increased and the Firmicutes PRA was decreased significantly in the EG group than those in the CG group (P < 0.05). At the genus level, similar to the total quantified proteins (Fig. 3d), the members of Prevotella were also the dominant proteins in DEPs (Fig. 3f), and the other abundant proteins were Clostridium, Bacteroides, Flavonifractor, and Ruminococcus (Fig. 3f).
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was used to explore the biological pathways for DEPs between the rumen fluid microbiota of EG and CG. Fig. 5a showed the KEGG pathway enrichment analysis of all DEPs. Fig. 5b showed the KEGG pathway enrichment analysis of all up-regulated DEPs, diet supplemented with CrPyr significantly enriched proteins involved in environmental information processing (HIF-1 signaling pathway, two-component systems); genetic information processing (ribosome, RNA degradation); human diseases (alzheimer disease, tuberculosis); metabolism (2-oxocarboxylic acid metabolism, biosynthesis of antibiotics, butanoate metabolism, carbon fixation in photosynthetic organisms, carbon metabolism, glycine, serine and threonine metabolism, glycolysis/gluconeogenesis, purine metabolism, pyruvate metabolism); organismal systems (longevity regulating pathway-worm). Fig. 5c showed the KEGG pathway enrichment analysis of all down-regulated DEPs, diet supplemented with CrPyr significantly decreased proteins involved in genetic information processing (ribosome, RNA polymerase); metabolism (alanine, aspartate and glytamate metabolism, arginine biosynthesis, biosynthesis of antibiotics, butanoate metabolism, carbon fixation in photosynthetic organisms, carbon fixation pathway in prokaryotes, carbon metabolism, citrate cycle (TCA cycle), fructose and mannose metabolism, glycine, serine and threonine metabolism, glycolysis/gluconeogenesis, glyoxylate and dicarboxylate metabolism, nitrogen metabolism, pentose and glucuronate interconversions, pyruvate metabolism, valine, leucine and isoleucine degradation); organismal systems (GABAergic synapse, glutamatergic synapse).
Interactive Pathways Explorer (IPath) analysis was also used to visualize the mutual relationship of DEPs in metabolic (Fig. 6a) and microbial metabolism (Fig. 6b), on which red lines show up-regulated pathways, green lines show down-regulated pathways, blue lines show both up-regulated and down-regulated pathways. As shown in metabolic and microbial metabolism, the up-regulated pathways mainly including lipid metabolism, glycolysis/gluconeogenesis, and pyruvate metabolism.
3.6. Administration of CrPyr affected core enzymes related to key metabolism
3.6.1. Fatty acid metabolism
As shown in Fig. 7a and Additional file 6: Table S5, dietary supplemented with CrPyr up-regulated the Acyl-CoA dehydrogenase (EC: 1.3.8.1) from Lachnoclostridium sp., Lachnospiraceae bacterium, and Oscillibacter sp. but down-regulated this enzyme from Muribaculum. Exposure to CrPyr up-regulated the Acetyl-CoA C-acetyltransferase (ACAA) (EC: 2.3.1.9) from Oscillibacter sp.. Moreover, the 3-oxoacyl-[acyl-carrier-protein] synthase 2 (EC: 2.3.1.179) from Lentimicrobiaceae bacterium was up-regulated, and the 3-oxoacyl-[acyl-carrier-protein] reductase (EC: 1.1.1.100) from Prevotella sp. BP1-148 was down-regulated. The description of DEPs involved in fatty acid metabolism were showed in Additional file 6: Table S5.
3.6.2. Pyruvate metabolism
As shown in Fig. 7b, for the pyruvate metabolism pathway, 62 DEPs were identified in the EG compared with the CG, of which 47 proteins were up-regulated and 15 proteins were down-regulated. Of the 47 up-regulated proteins, most were pyruvate, phosphate dikinase (PPDK) (EC: 2.7.9.1) (17). Others were pyruvate:ferredoxin (Flavodoxin) oxidoreductase (6) and pyruvate-flavodoxin oxidoreductase (2) (EC: 1.2.7.1), phosphoenolpyruvate carboxykinase (ATP) (7) (EC: 4.1.1.49), and so on. The description of DEPs involved in pyruvate metabolism were showed in Additional file 7: Table S6.
3.6.3. Glycolysis / Gluconeogenesis
As shown in Fig. 7c, for the glycolysis / gluconeogenesis, 101 DEPs were identified in the EG compared with the CG, of which 75 proteins were up-regulated and 26 proteins were down-regulated. Except for EC: 1.2.7.1, EC: 4.1.1.49, EC: 1.2.7.11, which were also involved in pyruvate metabolism, the most up-regulated protein was glyceraldehyde-3-phosphate dehydrogenase (21, including 3 Type I glyceraldehyde-3-phosphate dehydrogenase (Fragment) ) (EC: 1.2.1.12). Others were fructose-1,6-bisphosphate aldolase, class II and its isozyme (7) and (EC: 4.1.2.13), phosphoglycerate kinase (5) (EC: 2.7.2.3), pyrophosphate--fructose 6-phosphate 1-phosphotransferase (4) (EC: 2.7.1.90), enolase (4) (EC: 4.2.1.11), and so on. The description of DEPs involved in glycolysis / gluconeogenesis were showed in Additional file 8: Table S7.
3.6.4. Citrate cycle (TCA cycle)
As shown in Fig. 7d, for the TCA cycle, 39 DEPs were identified in the EG compared with the CG, of which 29 proteins were up-regulated and 10 proteins were down-regulated. Except for EC: 1.2.7.1, EC: 1.2.7.11, EC: 4.1.1.49, EC: 6.4.1.1, which also involved in pyruvate metabolism, the most up-regulated protein was succinate dehydrogenase/fumarate reductase and its isozyme (4) (EC: 1.3.5.4; EC: 1.3.5.1). The description of DEPs involved in TCA cycle were showed in Additional file 9: Table S8.
3.6.5. Nitrogen metabolism and biosynthesis of amino acids
As shown in Fig. 7e, for the nitrogen metabolism pathway, 28 DEPs were identified in the EG compared with the CG, of which 11 proteins were up-regulated and 17 proteins were down-regulated. Of the 11 up-regulated proteins, including 8 glutamate dehydrogenase (GDH) (EC: 1.4.1.4) and 3 glutamine synthetase (GS) (EC: 6.3.1.2). As shown in Fig. 7f, for the biosynthesis of amino acids, 94 DEPs were identified in the EG compared with the CG, of which 58 proteins were up-regulated and 36 proteins were down-regulated. The description of DEPs involved in nitrogen metabolism and biosynthesis of amino acids were showed in Additional file 10: Table S9, S10.