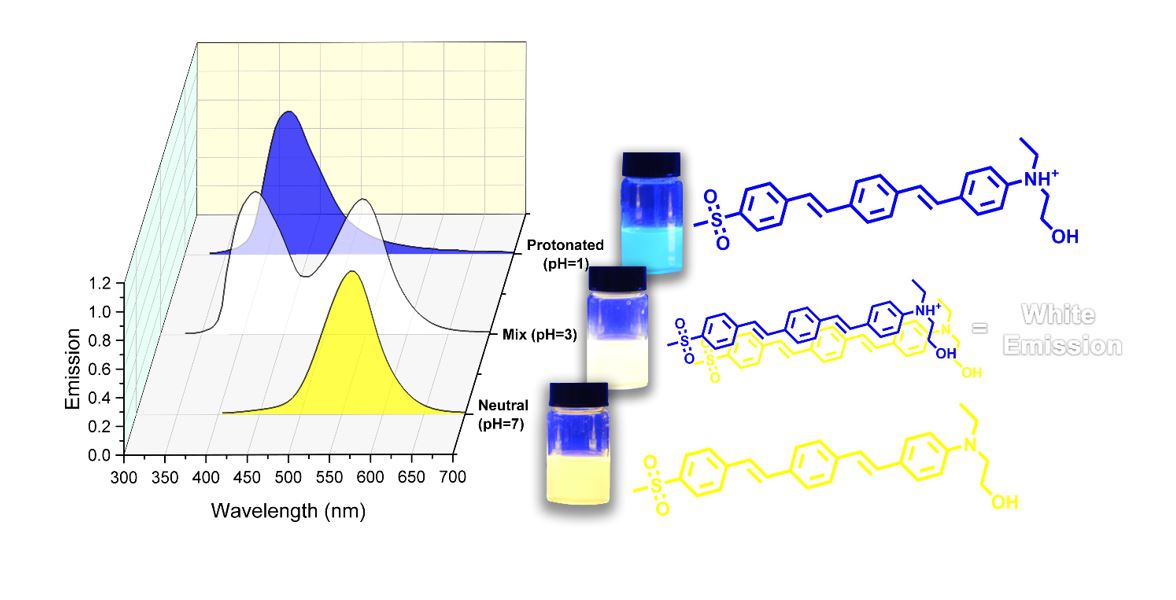
Highly efficient single-component white light emitters (SWLEs), are attractive candidates for the simple and cost-effective fabrication of high-performance lighting devices. This study introduced a donor–π–acceptor and a donor–π–donor stilbene-based chromophores, representing pH-responsive fluorescence. The emitters showed yellow and green fluorescence in their neutral form. At the same time, protonation of the chromophores caused blue fluorescence color with a strong hypsochromic shift. The white light emission (WLE) for these chromophores was observed at approximately pH=3 due to the simultaneous presence of the neutral and protonated forms of the chromophores, covering almost all the emission spectra in the visible region (400-700 nm). These chromophores presented exceptional white light quantum yields (Φ) between 31-54%, which was desirable for producing white light-emitting devices. Density functional theory (DFT) and time-dependent (TD)-DFT were applied to study the structural and electronic properties of the chromophores.
Research Article
New White Light-Emitting Halochromic Stilbenes with Remarkable Quantum Yields and Aggregation-Induced Emission
https://doi.org/10.21203/rs.3.rs-1053023/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 11 Feb, 2022
You are reading this latest preprint version
photophysical properties
white light emitters
halochromic materials
fluorescence
pH-sensitive
Light sources based on white organic light-emitting diodes (WOLEDs), owing to their high efficiency and flexibility, low final price, long lifespan, and good color quality have emerged as a powerful platform to replace the traditional lighting devices such as the incandescent bulb, fluorescent lamps, and in organic light-emitting diodes (LED).1–4 Conventional white light emission (WLE) in WOLEDs is obtained by combining three primary red, green, and blue emitters or two complementary-color emitters (for example, a yellow and a blue emitter).5–7 However, employing multiple emitters in fabricating WOLEDs is usually accompanied by several challenges such as complicated fabrication methods. Moreover, it is highly problematic to control the emission hue due to the energy transfer between different emitters. Additionally, the difference in photo-stability of emitters causes hue alteration in the long-term application of lighting devices.8–12 Employing single-component white light emitters (SWLEs) whose emission spectra cover the whole visible region ranging from 400 to 700 nm can be an exceptional alternative for the simple fabrication of WOLEDs without the previously mentioned drawbacks. The sole structure of single-component white light emitters (SWLEs) can eliminate charge transfer and stability problems. Moreover, SWLE-based lighting devices can enjoy lower fabrication costs and less driving voltage than traditional lighting sources.13–15 White fluorescence can be obtained through different photochemical procedures such as excited-state intramolecular proton transfer (ESIPT),16–19 aggregation-induced emission (AIE),20–22 halochromism22–30 or their combinations.31–33 Halochromism is a specific type of ionochromism in which a change in pH value can induce the color of a fluorescent compound. White light emission can be generated from pH-dependent fluorescent dyes at a specific pH where the emission of neutral and protonated forms is complementary and identical. The synthesis of halochromic chromophores is usually straightforward because these compounds are comparatively small molecules with a simple chemical structure.34 Moreover, tunable fluorescent devices can be easily fabricated by a single halochromic chromophore through pH regulation instead of using multiple emitters in separate layers. They also benefit from better colorfastness during multiple voltage cycles and prolonged application time.35 Some other applications of halochromic fluorescent chromophores include pH-sensitive sensors, biomedical probes, and textile dyes.36–44 However, only a limited number of halochromic white light emitters have been synthesized so far, and the low quantum yield is still a critical issue. Herein, in continuation of our previous reports,45–50 two highly efficient donor–π–acceptor and donor–π–donor stilbene-based fluorescent chromophores (Figure 1) with white light emission introduced and their photophysical properties investigated.
Compounds ASDSB and AADSB were synthesized based on our previously reported procedure in the literature.51
The solvatochromic effect of the chromophores was investigated from the absorption and emission spectra in low to high polarity solvents including xylene, ethyl acetate (ETA), dichloromethane (DCM), ethanol, and dimethylformamide (DMF). The corresponding results are shown in Figure 2 and Table 1.
Table 1. Optical data of the chromophores
|
Solvent |
λab (nm) |
λem (nm) |
Stokes shift (cm-1) |
ε (L.M-1.cm-1) |
E (eV)a |
Φb |
|
|
ASDSB |
Xylene |
394 |
505 |
5579 |
9300 |
2.69 |
0.73 |
|
DCM |
394 |
563 |
7619 |
65200 |
2.66 |
0.13 |
|
|
ETA |
394 |
554 |
7330 |
62700 |
2.69 |
0.1> |
|
|
DMF |
401 |
575 |
7546 |
61300 |
2.63 |
0.1> |
|
|
Ethanol |
394 |
570 |
7837 |
23300 |
2.66 |
0.1> |
|
|
|
|
|
|
|
|
|
|
|
AADSB |
Xylene |
391 |
451 |
3403 |
3400 |
2.83 |
0.55 |
|
DCM |
394 |
475 |
4523 |
49600 |
2.82 |
0.56 |
|
|
ETA |
392 |
471 |
4344 |
46000 |
2.82 |
0.52 |
|
|
DMF |
404 |
502 |
5655 |
61200 |
2.64 |
0.34 |
|
|
Ethanol |
389 |
477 |
4611 |
20400 |
2.82 |
0.68 |
a Calculated from the absorption spectra by using the empirical formula of
E(eV) = hc/λonset = 1240 (eV nm)/λonset(nm).
b 9,10-Diphenylanthracene was used as standard (Ф = 0.90 in cyclohexane).
The ASDSB and AADSB compounds showed a broad and structure-less absorption band ranging from 300 to 500 nm with a maximum peak at around 400 nm assigned to the intramolecular charge transfer (ICT) process from the amine and/or sulfonyl group. The absorption band of the chromophores revealed a negligible solvatochromic effect by increasing solvent polarity from low toward high polarity solvents. These observations illustrated that the chromophores exhibit low dipole moments at their ground state. Chromophore ASDSB showed a strong fluorescent emission in low to medium polarity solvents (i.e., xylene, DCM, and ETA) and a weak fluorescent emission in high polar solvents (i.e., ethanol and DMF). At the same time, AADSB showed intense fluorescent emission in all solvents. Chromophore ASDSB experienced a bathochromic shift from 505 nm in xylene to 575 nm in DMF, and its fluorescent color intensely changed from greenish-blue to orange. However, this trend was less pronounced for chromophore AADSB. This chromophore underwent a bathochromic shift from 451 nm in xylene to 502 in DMF. AADSB showed a blue fluorescent color in low polar media that was gradually changed to green with increasing solvent polarity. These results showed that the excited state of the chromophores is more polar and had more ICT characteristics than that of their ground state. The stabilization of the excited state by solvents with more polarity was the reason for the observed solvatochromism.52,53 The more distinct solvatochromic effect for chromophore ASDSB compared to that of chromophore AADSB may arise from the difference in their chemical structure. ASDSB with strong donor (amine) and acceptor (sulfonyl) substitutions exhibit more intense ICT characters compared to that of AADSB. These properties can result in the larger dipole moment and smaller energy band gap of ASDSB. Therefore, the compound at its excited state can easily stabilize by polar solvent molecules interactions. An excellent fluorescence quantum yield (Φ) with a value of 0.73 was recorded for the chromophore ASDSB. However, the fluorescence was significantly decreased with increasing solvent polarity from xylene to DMF corresponding to the non-radiative relaxation process. As mentioned earlier, polar solvents can stabilize the excited state of the fluorescent compound, narrow the energy gap between its ground and excited state, and induce radiation less decay.54–56 In the case of chromophore AADSB, the fluorescence quantum yield (Φ) was found to be high in all solvents with a minimum value of 0.34 in DMF and a maximum value of 0.68 in ethanol.
The solvent effects on fluorescence characteristic of a molecule can be demonstrated by Lippert-Mataga (LM) formalism57:

In this equation, Δv , Δµ, h, c, a, Δf, ε and n are Stocks shift, the difference of ground state and excited state dipole moments, Planks constant, speed of light, the radius of cavity, solvent polarizability, dielectric constant and refractive index respectively. Equation (2) shows the re-orientation of solvent molecules with dielectric constant (ε) and refractive index (n). The Lippert-Mataga plot is shown in Figure 3. The slope of the fitted line on the data points indicates the sensitivity of the fluorescence to the solvent polarity. The higher slope of ASDSB compared with AADSB (8305 and 6131, respectively) shows that the difference of dipole moment of the ASDSB in the excited state and ground state is higher than the AADSB due to the asymmetric structural design. This result explains the higher stokes shift and lower quantum yields (Φ) of ASDSB in polar solvents.
The absorption and emission spectra of ASDSB and AADSB in DCM upon addition various amounts of trifluoroacetic acid (TFA) were recorded to investigate the effect of protonation on photophysical properties of the chromophores and the results were shown in Figure 4.
As seen, both absorption and emission spectra of the chromophores showed minor pH responsivity with the variation of pH from 7 to 4. A significant hypsochromic shift was observed for the absorption and emission bands of the chromophores with further decreasing pH from 4 to 1. Correspondingly, the color of the solutions was changed from yellow to colorless for ASDSB and green to blue for AADSB with decreasing pH. The fluorescent hue was also changed from yellow to blue for ASDSB solutions and green to blue for AADSB solutions after protonation of the chromophores. The observed hypsochromic shift for chromophores ASDSB and AADSB were 63 nm (from 397 nm at pH=7 to 334 nm at pH=1) and 44 nm (from 394 nm at pH=7 to 350 nm at pH=1) for their absorption band, respectively. These values were found to be 134 nm (from 563 nm at pH=7 to 429 nm at pH=1), and 57 nm (from 475 nm at pH=7 to 418 nm at pH=1) for their emission band, respectively. These results can be originated from the protonation of the chromophores with increasing acid concentration interrupting the ICT process. In fact, the protonation of the strong electron-donating substitution on the chromophore's molecular structure in acidic conditions can turn the amine group to quaternary ammonium salt as a strong electron-withdrawing group. This procedure weakens electron-donating ability and interrupts the chromophore's push-pull structure, leading to a less ICT character.58,59
The most surprising aspect of the data in Figure 4 and Table 2 is that the chromophores can illustrate white light emission at pH around 3 with the commission international de l'eclairage(CIE 1931) color coordinates of (0.33, 0.32) for ASDSB and (0.33, 0.31) for AADSB, respectively. This relatively rare phenomenon in fluorescent compounds stemmed from the pH sensitivity of the chromophores. In other words, at a specific pH where the identical amount of neural and protonated forms of the compounds are simultaneously present in their solutions, white light fluorescence can be observed since the emission bands are broad and complementary colors. Another striking observation to emerge from the data was compelling white light quantum yield (Φ) with the values of 0.31 and 0.54 for chromophore ASDSB and AADSB, respectively. It was reported that lighting accounts for almost 20% of the total electricity produced in the globe.60 Only a small increase in quantum yield of white light emitters could substantially decline electricity consumption. Therefore, the observed results highlighted the potential application of ASDSB and AADSB as remarkable options in fabricating lighting sources.
Table 2. Optical data of the chromophores at pH = 3
|
Chromophore |
Solvent |
(x,y) |
Φ |
|
ASDSB |
DCM |
(0.33, 0.32) |
0.31 |
|
AADSB |
DCM |
(0.33, 0.31) |
0.54 |
The emission spectra of the chromophores in the mixture of DMF (good solubility of chromophores) and water (low solubility of chromophores) were obtained to evaluate AIE properties for these compounds. The corresponding results are depicted in Figure 5.
As can be seen, despite the relative similarity in the molecular structure of chromophore ASDSB and AADSB, they represented different aggregation characteristics. For chromophore ASDSB, upon increasing water fraction from 0 to 70%, the intensity of the emission band at 575 nm was steadily decreased, accompanied by the disappearance of the shoulder at 473 nm. The observed color change from orange to red may be attributed to stabilizing dipole moment at the ground state by increasing solvent interactions such as hydrogen bonding. A slight increase in the emission intensity was observed with further increasing water fractions from 70 to 90%, showing the aggregation-induced emission (AIE) phenomenon for chromophore ASDSB. These results may originate from the restriction of fluorophore intramolecular motions due to the formation of molecular aggregations due to low solubility in the mixture solvent. Chromophore AADSB displayed a gradual rise in the emission intensity with the concomitant red shift from 502 nm to 513 nm as the water fraction increased from 0 to 60%. Following the increase of water fraction to 70%, a significant drop in the emission intensity was observed for this sample. Finally, the fluorescence was quenched by further increasing water fraction to 90%, indicative of aggregation-caused quenching (ACQ) for AADSB. The increase in the fluorescence of chromophore ASDSB (after addition of more than 70% water) and AADSB (up to 60% water portion in the solvents mixture) can be explained by the aggregate formation of the chromophores in poor solvents restricting the intermolecular rotations.61,62 These results were representative of the AIE character in chromophore ASDSB and AADSB.
Quantum mechanical studies
Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) were chosen as the most practical ways to investigate the chromophores' electronic structure and vertical transition energies.
Figure 6 shows the resulting optimized geometries of ASDSB and AADSB employingB3LYP/ 6-311+g(2d,p) in a DCM solvent environment. The side view of both molecules shows a planar geometry of the three phenyl rings. However, the bulkiness of the amine moiety induces some degree of dihedral twist between the two adjacent phenyl rings. Therefore, in agreement with the higher Stokes shift of ASDSB in experiments, the backbone of ASDSB is supposed to be more rigid than AADSB. The frontier orbitals of the molecules are shown in Figure 6 (c) and (d). The highest occupied molecular orbital (HOMO) of the ASDSB is located on amine and the two adjacent phenyl rings. At the same time, the electrons in the HOMO of the AADSB are distributed on the entire backbone. While the lowest unoccupied molecular orbital (LUMO) in AADSB is somehow localized on the phenyl rings, the LUMO of ASDSB is localized on the electron-withdrawing sulfonyl group. The push-pull electronic transfer in ASDSB brought a deeper HOMO and LUMO than AADSB (Table 3). A most remarkable result from the data is that the HOMO to LUMO transformation in ASDSB and AADSB is almost different, which suggests different band gaps and absorption wavelengths (λmax). Still, according to the experiment and the TD-DFT simulation (Table 3), both molecules show the same λmax. On the other hand, molecular electrostatic potential (MEP) surfaces and vectors of dipole moment (Figure 6 (e) and (f)) illustrate that the ASDSB compared with the ASDSB, has a more polar structure. In other words, replacing the electron- withdrawing sulfonyl group with an electron-donating amine grope has no meaningful impact on the absorption characteristics. This is explained by the long system of conjugation, which is brought by the sequence of stilbenes in the backbone of the molecules. Putting the donor and acceptor in such a far distance led to the same length of conjugation for both molecules and diminishing the push-pull effect on the absorption wavelength, yet different solvatochromic and fluorescence characteristics, as seen in experiments and Lipppert-Mataga study.
Table 3. Ground state (DFT) and excited state (TD-DFT) calculated parameters of ASDSB and AADSB.
|
|
ASDSB |
AADSB |
|
Ground state |
||
|
HOMO |
-5.16 |
-4.90 |
|
LUMO |
-2.39 |
-1.92 |
|
Gap |
2.78 (2.66) |
2.98 (2.84) |
|
Dipole moment |
11.7 |
3.1 |
|
Excited-state |
||
|
λmax(nm) |
404 (394) |
401 (394) |
|
Oscillator strength |
2.3772 |
2.5841 |
|
Major contributions |
HOMO → LUMO (78%) |
HOMO → LUMO (89%) |
In conclusion, two stilbene-based chromophores were introduced, which showed yellow and green emission fluorescence in their neutral form and blue fluorescence color with a strong hypsochromic shift in the protonated forms. These chromophores showed WLE at pH=3 due to the simultaneous presence of the neutral and protonated chromophores, representing pH-responsive fluorescence. Remarkable white light quantum yields (Φ) were observed for producing white light-emitting devices in these chromophores, suggesting a high potential application of the compounds in the preparation of high-performance lighting devices.
General. Chemicals were purchased from Fluka and Aldrich companies and used without further purification. The known products were characterized by comparing their spectral and physical data with those reported in the literature. 1H NMR (250 MHz) and 13C NMR (62.5 MHz), spectra were recorded on a Brucker (250 MHz) Avance DRX. FT-IR spectroscopy (Shimadzu FT-IR 8300 spectrophotometer), were employed for the characterization of the products. Melting points were determined in open capillary tubes in a Buchi melting point B-545. The reaction monitoring was accomplished by TLC on silica gel PolyGram SILG/UV254 plates. Column chromatography was carried out on columns of silica gel 60 (70−230 mesh).
Procedure for the synthesis of compounds ASDSB and AADSB
1-(Methylsulfonyl)-4-(4-vinylstyryl)benzene (C). A sealed Schlenk tube was charged with1-bromo-4-methanesulfonyl-benzene (A; 5 mmol, 1.17 g), and K2CO3 (10.0 mmol, 1.4 g), Pd(OAc)2 (1.2 mol%, 14 mg), DPEPhos ligand (2.4 mol%, 65 mg) and it was evacuated and backfilled with argon. Then 1,4-divinyl-benzene (B; 5 mmol, 0.6 mL) and 5 mL of dry DMF was added to the reaction mixture under fellow of argon and tube was sealed with a screw-cap and the resulting mixture was heated in an oil bath at 120°C for 6h. To obtain the pure product its was purified by column chromatography (hexane/ethyl acetate: 10/1) (1.04g, 73%). Yellow solid; mp 182.7°C. IR (KBr): 3448, 3016, 1589, 1412, 1311, 1149, 957, 833, 764, 448 cm-1. 1H-NMR (250 MHz, CDCl3/TMS) δ (ppm): 3.07 (s, 3H), 5.29 (d, J = 11.0 Hz, 1H), 5.80 (d, J = 17.5 Hz, 1H), 6.73 (dd, J = 17.6, 10.7 Hz, 1H), 7.12 (d, J = 16.5, 1H), 7.24 (d, J = 16.5 Hz, 1H), 7.43 (d, J = 8.3 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.67 (d, J = 8.5 Hz, 2H), 7.92 (d, J = 8.5 Hz, 2H). 13C-NMR (62.5 MHz, CDCl3/TMS) δ (ppm): 44.6, 114.5, 126.3, 126.7, 127.0, 127.1, 127.9, 132.2, 135.7, 136.2, 137.9, 142.8. Anal. Cal. C17H16O2S (284.4): C, 71.80; H, 5.67; O, 11.25; S, 11.28; found: C, 71.85; H, 5.71.
2-4- 2-{Ethyl-[4-(2-{4-[2-(4-methanesulfonyl-phenyl)-vinyl]-phenyl}-vinyl)-phenyl]-amino}-ethanol (ASDSB). A sealed Schlenk tube was charged with compound C (1 mmol, 0.29 g), 2-[Ethyl-(4-iodo-phenyl)-amino]-ethanol (D; 1 mmol, 0.29 g), and K2CO3 (2.5 mmol, 0.35 g), Pd(OAc)2 (1.2 mol%, 2.7 mg), DPEPhos ligand (2.4 mol%, 13 mg) and it was evacuated and backfilled with argon. Then 5 mL of dry DMF was added to the reaction mixture under fellow of argon and tube was sealed with a screw-cap and the resulting mixture was heated in an oil bath at 120°C for 12h. After completion of the reaction, the mixture was filtered (in hot form) and the remaining solid was washed with DMF (2 mL) in order to separate the catalyst. Subsequently, water (10 mL) was added to the solution in order to precipitate product. The obtained solid was purified by column chromatography (hexane/ethyl acetate: 10/2) to obtain the pure product (0.4g, 90%). Orange solid; mp 275.5°C.1 IR (KBr): 3300, 3000, 1600, 1590, 1510, 1400, 1350, 1290, 1180, 1130, 1080, 1060, 830, 820, 760, 550 cm-1. 1H-NMR (250 MHz, DMSO-d6/TMS) δ (ppm): 1.07 (t, J = 6.8 Hz, 3H), 3.20 (s, 3H), 3.32-3.51 (m, 6H), 4.72 (s, 1H), 6.66 (d, J = 8.6 Hz, 2H), 6.93 (d, J = 16.4 Hz, 2H), 7.15 (d, J = 16.4 Hz, 2H), 7.36-7.42 (m, 2H), 7.48-7.62 (m, 6H), 7.81-7.90 (m, 2H). Anal. Cal. C27H29NO3S (447.6): C, 72.45; H, 6.53; N, 3.13; O, 10.72; S, 7.16; found: C, 72.51; H, 6.58; N, 3.19.
2-[Ethyl-(4-{2-[4-(2-{4-[ethyl-(2-hydroxy-ethyl)-amino]-phenyl}-vinyl)-phenyl]-vinyl}-phenyl)-amino]-ethanol (AADSB). A sealed Schlenk tube was charged with 2-[Ethyl-(4-iodo-phenyl)-amino]-ethanol (D; 2 mmol, 0.60 g), K2CO3 (5 mmol, 0.68 g), Pd(OAc)2 (1.2 mol%, 5.5 mg), DPEPhos ligand (2.4 mol%, 26 mg) and it was evacuated and backfilled with argon. Then 1,4-divinyl-benzene (B; 1 mmol, 0.12 mL) and 6 mL of dry DMF wereadded to the reaction mixture under the argon atmosphere. The tube was sealed with a screw-cap, and the resulting mixture was heated in an oil bath at 120°C for 6h. The reaction was followed by TLC. After completion of the reaction, the mixture was cooled down to room temperature and filtered. The remaining solid was washed with dichloromethane (3 x 5 mL) to separate the catalyst. After the extraction of dichloromethane from water, the organic extract was dried over Na2SO4. The products were purified by column chromatography (hexane/ethyl acetate: 10/2) to obtain the pure product (0.4g, 88%). Yellow solid; mp 216.5°C. IR (KBr): 3389, 2922, 1601, 1520, 1360, 1267, 1180, 1051, 964, 823, 550 cm-1. 1H-NMR (250 MHz, DMSO-d6) )δppm): 1.04-1.14 (m, 6H), 3.31-3.53 (m, 12H), 4.70 (m, 2H), 6.55-6.67 (m, 4H), 6.85-7.10 (m, 8H), 7.22-7.66 (m, 4H). M/z (%): 456 (94.5%, (M)+). Anal. Cal. C30H36N2O2 (456.6): C, 78.91; H, 7.95; N, 6.13; O, 7.01; found: C, 78.98; H, 7.99; N, 6.18.
Theoretical calculations
The geometry of the molecules in the ground state was optimized by the first-principles density functional theory (DFT). The calculations carried byB3LYP functional63 and 6-311+g(2d,p)as the basis set. The most stable geometry was found by examining different isomers and configurations and examining vibrational frequency calculation. The excited state of the molecules was simulated by time-dependent density functional theory(TD-DFT). The model was the Coulomb attenuating employing the B3LYP hybrid functional (CAM-B3LYP)64 and the same basis-set as the DFT method. The Polarizable Continuum Model with the integral equation formalism (IEFPCM)was chosen to model the dichloromethane (DCM) as solvent. Calculations were carried out using Gaussian 09.65
Author contributions
The work was conceptualized by FP and synthesized the organic compounds. AM and SG performed the photophysical experiments. AA and FE performed the computational analysis. The manuscript was prepared and edited by all the authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
Financial support from the research councils of Shiraz University and Amirkabir University of Technology are gratefully acknowledged.
- Gather, M. C., Köhnen, A. & Meerholz, K. White organic light-emitting diodes. Advanced Materials 23, 233–248 (2011).
- Yin, Y., Ali, M. U., Xie, W., Yang, H. & Meng, H. Evolution of white organic light-emitting devices: from academic research to lighting and display applications. Mater. Chem. Front. 3, 970–1031 (2019).
- Bernal, W. et al. White Organic Light emitting diodes based On exciplex states by using a new carbazole derivative as single emitter Layer. Dyes and Pigments 163, 754–760 (2019).
- Wei, X. et al. A new strategy for structuring white organic light-emitting diodes by combining complementary emissions in the same interface. J. Mater. Chem. C 8, 2772–2779 (2020).
- Zhang, H., Chen, S. & Sun, X. W. Efficient Red/Green/Blue Tandem Quantum-Dot Light-Emitting Diodes with External Quantum Efficiency Exceeding 21%. ACS Nano 12, 697–704 (2018).
- Maiti, D. K., Bhattacharjee, R., Datta, A. & Banerjee, A. Modulation of Fluorescence Resonance Energy Transfer Efficiency for White Light Emission from a Series of Stilbene-Perylene Based Donor–Acceptor Pair. The Journal of Physical Chemistry C 117, 23178–23189 (2013).
- Peng, C. et al. Efficient and chromaticity-stable flexible white organic light-emitting devices based on organic–inorganic hybrid color-conversion electrodes. RSC Adv. 9, 22577–22585 (2019).
- Pati, A. K., Gharpure, S. J. & Mishra, A. K. White Light Emission in Butadiyne Bridged Pyrene–Phenyl Hybrid Fluorophore: Understanding the Photophysical Importance of Diyne Spacer and Utilizing the Excited-State Photophysics for Vapor Detection. The Journal of Physical Chemistry A 120, 5838–5847 (2016).
- Xie, Z. et al. White-Light Emission Strategy of a Single Organic Compound with Aggregation-Induced Emission and Delayed Fluorescence Properties. Angewandte Chemie International Edition 54, 7181–7184 (2015).
- Yang, Q.-Y. & Lehn, J.-M. Bright white-light emission from a single organic compound in the solid State. Angewandte Chemie International Edition 53, 4572–4577 (2014).
- Jin, X.-H., Chen, C., Ren, C.-X., Cai, L.-X. & Zhang, J. Bright white-light emission from a novel donor–acceptor organic molecule in the solid state via intermolecular charge transfer. Chemical Communications 50, 15878–15881 (2014).
- Chen, C. et al. Photo-facilitated aggregation and correlated color temperature adjustment of single component organic solid state white-light emitting materials. Journal of Materials Chemistry C 3, 4563–4569 (2015).
- Wang, L. et al. Highly efficient white organic light-emitting diodes with single small molecular emitting material. Applied physics letters 91, 183504 (2007).
- Mazzeo, M. et al. Bright White Organic Light-Emitting Devices from a Single Active Molecular Material. Advanced Materials 17, 34–39 (2005).
- Chen, Z., Ho, C.-L., Wang, L. & Wong, W.-Y. Single-Molecular White-Light Emitters and Their Potential WOLED Applications. Advanced Materials 32, 1903269 (2020).
- Cheng, J. et al. A Single 2-(2′-Hydroxyphenyl) benzothiazole Derivative Can Achieve Pure White-Light Emission. Chemistry–An Asian Journal 9, 3215–3220 (2014).
- Li, B. et al. Dual-emissive 2-(2′-hydroxyphenyl) oxazoles for high performance organic electroluminescent devices: discovery of a new equilibrium of excited state intramolecular proton transfer with a reverse intersystem crossing process. Chemical science 9, 1213–1220 (2018).
- Zhao, J., Ji, S., Chen, Y., Guo, H. & Yang, P. Excited state intramolecular proton transfer (ESIPT): from principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Physical Chemistry Chemical Physics 14, 8803–8817 (2012).
- Duarte, L. G. T. A. et al. White-light generation from all-solution-processed OLEDs using a benzothiazole–salophen derivative reactive to the ESIPT process. Phys. Chem. Chem. Phys. 21, 1172–1182 (2019).
- Molla, M. R. & Ghosh, S. Hydrogen-bonding-mediated J-aggregation and white-light emission from a remarkably simple, single-component, naphthalenediimide chromophore. Chemistry-A European Journal 18, 1290 (2012).
- Bhattacharya, S. & Samanta, S. K. Unusual Salt-Induced Color Modulation through Aggregation-Induced Emission Switching of a Bis-cationic Phenylenedivinylene-Based π Hydrogelator. Chemistry–A European Journal 18, 16632–16641 (2012).
- Sharma, C. P. et al. Synthesis of Solution-Processable Donor–Acceptor Pyranone Dyads for White Organic Light-Emitting Devices. The Journal of organic chemistry 84, 7674–7684 (2019).
- Li, M., Yuan, Y. & Chen, Y. Acid-induced multicolor fluorescence of pyridazine derivative. ACS applied materials & interfaces 10, 1237–1243 (2018).
- Täuscher, E. et al. Classical heterocycles with surprising properties: the 4-hydroxy-1, 3-thiazoles. Tetrahedron Letters 52, 2292–2294 (2011).
- Yamaguchi, K., Murai, T., Guo, J.-D., Sasamori, T. & Tokitoh, N. Acid-Responsive Absorption and Emission of 5-N-Arylaminothiazoles: Emission of White Light from a Single Fluorescent Dye and a Lewis Acid. ChemistryOpen 5, 434 (2016).
- Achelle, S., Rodríguez-López, J., Katan, C. & Robin-le Guen, F. Luminescence behavior of protonated methoxy-substituted diazine derivatives: toward white light emission. The Journal of Physical Chemistry C 120, 26986–26995 (2016).
- Huynh, H. V., He, X. & Baumgartner, T. Halochromic generation of white light emission using a single dithienophosphole luminophore. Chemical Communications 49, 4899–4901 (2013).
- Liu, D., Zhang, Z., Zhang, H. & Wang, Y. A novel approach towards white photoluminescence and electroluminescence by controlled protonation of a blue fluorophore. Chemical Communications 49, 10001–10003 (2013).
- Takahashi, M., Enami, Y., Ninagawa, H. & Obata, M. A novel approach to white-light emission using a single fluorescent urea derivative and fluoride. New Journal of Chemistry 43, 3265–3268 (2019).
- Chaudhary, J. et al. A New Aggregation Induced Emission Active Halochromic White Light Emissive Molecule: Combined Experimental and Theoretical Study. J. Phys. Chem. C 124, 15406–15417 (2020).
- Chen, Y. et al. Color-Tunable and ESIPT-Inspired Solid Fluorophores Based on Benzothiazole Derivatives: Aggregation-Induced Emission, Strong Solvatochromic Effect, and White Light Emission. ACS Appl. Mater. Interfaces 12, 55094–55106 (2020).
- Samanta, S., Manna, U. & Das, G. White-light emission from simple AIE–ESIPT-excimer tripled single molecular system. New J. Chem. 41, 1064–1072 (2017).
- Maity, A., Ali, F., Agarwalla, H., Anothumakkool, B. & Das, A. Tuning of multiple luminescence outputs and white-light emission from a single gelator molecule through an ESIPT coupled AIEE process. Chem. Commun. 51, 2130–2133 (2015).
- Mukherjee, S. & Thilagar, P. Organic white-light emitting materials. Dyes and Pigments 110, 2–27 (2014).
- Park, Y. I. et al. A new pH sensitive fluorescent and white light emissive material through controlled intermolecular charge transfer. Chemical science 6, 789–797 (2015).
- Yang, Y. et al. An organic white light-emitting fluorophore. Journal of the American Chemical Society 128, 14081–14092 (2006).
- Tydlitát, J. et al. Photophysical properties of acid-responsive triphenylamine derivatives bearing pyridine fragments: Towards white light emission. Dyes and Pigments 146, 467–478 (2017).
- Giovanella, U. et al. In Situ electroluminescence color tuning by thermal deprotonation suitable for thermal sensors and anti-fraud labels. ChemPhysChem 18, 2157–2161 (2017).
- Hu, Y. et al. Colorimetric and fluorescent detecting phosgene by a second-generation chemosensor. Analytical chemistry 90, 3382–3386 (2018).
- Shen, J. et al. Fabrication of smart pH-responsive fluorescent solid-like giant vesicles by ionic self-assembly strategy. The Journal of Physical Chemistry C 120, 27533–27540 (2016).
- Kumar, R., Yadav, R., Kolhe, M. A., Bhosale, R. S. & Narayan, R. 8-Hydroxypyrene-1, 3, 6-trisulfonic acid trisodium salt (HPTS) based high fluorescent, pH stimuli waterborne polyurethane coatings. Polymer 136, 157–165 (2018).
- Zhang, W., Zhang, Y., Cheng, Y., Qin, C. & Chen, G. A hemicyanine fluorescent reactive cationic dye: synthesis and applications on wool fabrics. Coloration Technology 131, 316–321 (2015).
- Nagarasu, P. et al. Structure controlled solvatochromism and halochromic fluorescence switching of 2,2′-bipyridine based donor–acceptor derivatives. New J. Chem. 44, 14421–14428 (2020).
- Siriwibool, S. et al. Near-Infrared Fluorescent pH Responsive Probe for Targeted Photodynamic Cancer Therapy. Scientific Reports 10, 1283 (2020).
- Mahmoodi, A., Panahi, F., Eshghi, F. & Kimiaei, E. A novel tetra-stilbene-based fluorescent compound: Synthesis, characterization and photophysical properties evaluation. Journal of Luminescence 199, 165–173 (2018).
- Karimi-Alavijeh, H., Panahi, F. & Gharavi, A. Photo-switching effect in stilbene organic field effect transistors. Journal of Applied Physics 115, 093706 (2014).
- Sharbati, M. T., Panahi, F. & Gharavi, A. Near-infrared organic light-emitting diodes based on donor-pi-acceptor oligomers. IEEE Photonics Technology Letters 22, 1695–1697 (2010).
- Sharbati, M. T., Panahi, F., Nekoei, A.-R., Emami, F. & Niknam, K. Blue to red electroluminescence emission from organic light-emitting diodes based on π-conjugated organic semiconductor materials. Journal of Photonics for Energy 4, 043599 (2014).
- Miri, F. S., Kandi, S. G. & Panahi, F. Photophysical Properties of a Donor-π-Acceptor Distyrylbenzene Derivative in Solution and Solid state. Journal of Fluorescence 1–10 (2020).
- Panahi, F., Mahmoodi, A., Ghodrati, S. & Eshghi, F. A novel donor–π–acceptor halochromic 2, 6-distyrylnaphthalene chromophore: synthesis, photophysical properties and DFT studies. RSC Advances 11, 168–176 (2020).
- Niknam, K., Gharavi, A., Nezhad, M. R. H., Panahi, F. & Sharbati, M. T. Synthesis of some new 1, 4-distyrylbenzenes using immobilized palladium nanoparticles on silica functionalized morpholine as a recyclable catalyst. Synthesis 2011, 1609–1615 (2011).
- Meisner, Q. J. et al. Fluorescence of hydroxyphenyl-substituted “click” triazoles. The Journal of Physical Chemistry A 122, 2956–2973 (2018).
- Carlotti, B. et al. Evaluation of hyperpolarizability from the solvatochromic method: Thiophene containing push–pull cationic dyes as a case study. The Journal of Physical Chemistry C 122, 2285–2296 (2018).
- Song, H. et al. Solvent Polarity Dependent Excited State Dynamics of 2′-Hydroxychalcone Derivatives. The Journal of Physical Chemistry C 122, 15108–15117 (2018).
- Maffeis, V., Brisse, R., Labet, V., Jousselme, B. & Gustavsson, T. Femtosecond Fluorescence Upconversion Study of a Naphthalimide–Bithiophene–Triphenylamine Push–Pull Dye in Solution. The Journal of Physical Chemistry A 122, 5533–5544 (2018).
- Zhang, Y. et al. Solvent effect and two-photon optical properties of triphenylamine-based donor–acceptor fluorophores. The Journal of Physical Chemistry C 119, 27630–27638 (2015).
- Nourmohammadian, F., Alikhani, M. Y., Gholami, M. D. & Abdi, A. A. Benzothiazole-Based Bis-azo Cationic Fluorescent Dyes with Extended Conjugated Systems: Synthesis and Properties. Journal of Applied Solution Chemistry and Modeling 4, 83–94 (2015).
- Dou, C., Han, L., Zhao, S., Zhang, H. & Wang, Y. Multi-Stimuli-Responsive Fluorescence Switching of a Donor−Acceptor π-Conjugated Compound. J. Phys. Chem. Lett. 2, 666–670 (2011).
- Tolosa, J., Solntsev, K. M., Tolbert, L. M. & Bunz, U. H. F. Unsymmetrical Cruciforms. J. Org. Chem. 75, 523–534 (2010).
- Wu, H., Ying, L., Yang, W. & Cao, Y. Progress and perspective of polymer white light-emitting devices and materials. Chem. Soc. Rev. 38, 3391–3400 (2009).
- Wang, Y. et al. Twisted Donor–Acceptor Cruciform Luminophores Possessing Substituent-Dependent Properties of Aggregation-Induced Emission and Mechanofluorochromism. J. Phys. Chem. C 122, 2297–2306 (2018).
- Vasu, A. K., Radhakrishna, M. & Kanvah, S. Self-Assembly Tuning of α-Cyanostilbene Fluorogens: Aggregates to Nanostructures. J. Phys. Chem. C 121, 22478–22486 (2017).
- Beck, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 98, 5648–6 (1993).
- Yanai, T., Tew, D. P. & Handy, N. C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chemical physics letters 393, 51–57 (2004).
- Frisch, M. et al. gaussian 09, Revision d. 01, Gaussian. Inc., Wallingford CT 201, (2009).
No competing interests reported.
- GraphicalAbstract.jpg
Halochromic stilbene-based chromophores showed white light emission at pH=3 due to the simultaneous presence of the neutral and protonated form of the chromophores, representing pH-responsive fluorescence with remarkable quantum yields (Φ). Farhad Panahi,* Ali Mahmoodi, Sajjad Ghodrati, Ali Ashtianib and Fazlolah Eshghi
{kind=link}