Patient samples
Three patient tissue samples from PCa patients not receiving chemotherapy, hormone therapy, or radiotherapy were obtained before surgery from Chosun University Hospital during 01 June 2020 and 31 July 2020 (Chosun University Hospital (CHOSUN 2020-06-001)). Tumours and normal tissues were harvested and immediately fixed in a formalin solution (neutral buffered, 10%; Sigma-Aldrich) for 24 h. Tissue specimens were successively dehydrated in ethanol and treated with xylene. Paraffin embedded tissues were sliced into 5 μm-thick sections, deparaffinised with xylene and rehydrated through graded alcohol solutions. The tissues were stained with H&E for histological analysis. Immuno-histochemical studies were performed using the NovolinkTM Polymer Detection System kit (Leica Biosystems) as per the manufacturer’s instructions. Antibodies specific for IL-6 (1:200; Cell Signaling Technology) and RANKL (1:200, Cell Signaling Technology) were used.
Cell lines and cultures
Human prostate adenocarcinoma luciferase-labelled PC3luc cells were obtained from Professor Park and maintained at 37°C/5% CO2 in Rowell Park Memorial Institute (RPMI) 1640 medium (Welgene, Korea) supplemented with 10% heat-inactivated foetal bovine serum (certified, GIBCO, USA) and a 10% antibiotic solution (Welgene).
Cloning of hRANKL
The RNA used to clone hRANKL cDNA was extracted from MG63 cells (ATCC® CRL-1427™) expressing RANKL. The quality of the extracted RNA was verified by agarose gel electrophoresis. The cDNA was prepared using the AccuPower RT PreMix Kit (Bioneer, Daejeon, Korea), according to the manufacturer’s instructions. Amplification and cloning of the hRANKL fragment were carried out in a reaction mixture comprising KOD polymerase buffer, 10 mM dNTPs, 25 mM magnesium chloride (MgCl2), 10 μM primers (hRANKL-BclI: 5'-TGATCAAAGCTTGAAGCTCAGCCTTTTGC-3' and hRANKL-XhoI: 5'-CTCGAGATCTATATCTCGAACTTTAAAAGCCCC-3'), 2.5 U of KOD DNA polymerase (EMD Millipore, Billerica, MA, USA) and 2 μL of the RANKL gene construct (template). The thermal cycling conditions were as follows: initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s and extension at 70°C for 30 s. The polymerase chain reaction (PCR) product was cloned into the BamH1/XhoI sites of a pMX vector (CELL BIOLABS, USA). Sequence analyses were carried out using programs in Vector NTI Advance 9.1.0 (Invitrogen, Carlsbad, CA, USA).
Retroviral hRANKL transduction
Plat-E cells were seeded at a density of 3 × 105 cells/well in a six-well plate for 24 h and then transiently transfected with hRANKL/pMX using 0.2 μg plasmid and 0.6 μL of the FuGENE HD transfection reagent (Promega, Madison, WI, USA), according to the manufacturer’s protocol. After incubation, the DNA/FuGENE mixture was added drop-wise onto Plat-E cells. Viral supernatants were recovered from the culture medium at 48 h after transfection. Virus-containing supernatants were filtered through 0.45 μm non-pyrogenic filters and supplemented with 10 μg/mL polybrene (Sigma-Aldrich).
Reverse-transcription quantitative PCR (RT-qPCR)
Total RNA was extracted from PCa cells using Trizol (Invitrogen) and 1 μg was used for RT-qPCR along with oligo-dT primers (10 μg) and dNTPs (10 mM). Next, qRT-PCR was performed to analyse cDNA using SYBR Green SuperMix (BIORAD, USA) on a CFX Connect Real-Time System (BIORAD, USA). All target gene primers were purchased from Bioneer Co. (Daejeon, Korea) and the cDNA was amplified using the following primer sets:
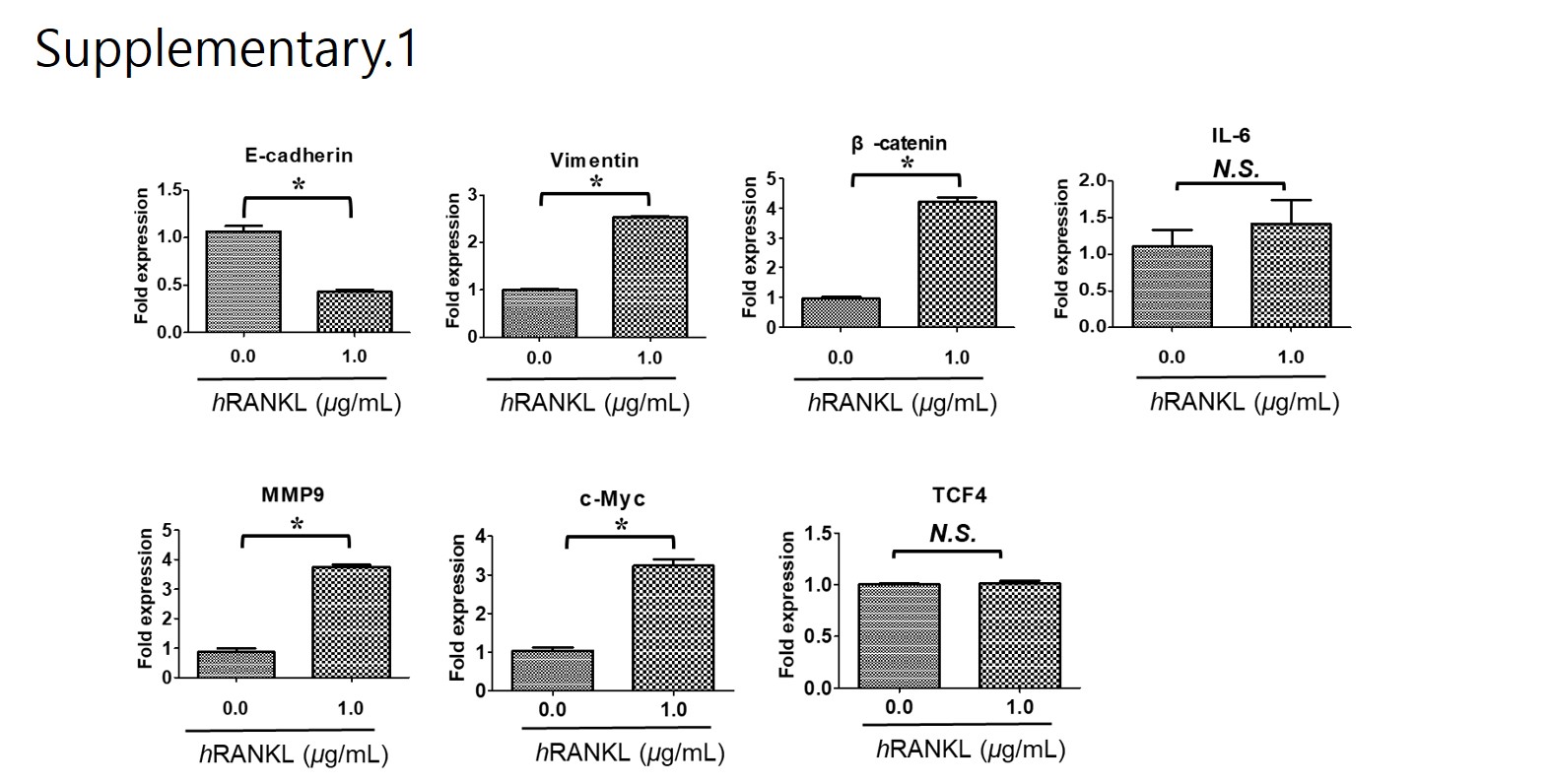
E-cadherin (h): 5'-TGGAGGAATTCTTGCTTTGC-3' (forward) and 5'-TGGAGGAATTCTTTTGC-3' (reverse); vimentin (h): 5'-GACGCCATCAACACCGAGTT-3' (forward) and 5'-GACGCCATC AACACCGAGTT-3' (reverse); β-catenin (h): 5'-ACAAACTGTTTTGAAAATCCA-3' (forward) and 5'-CGAGTCATTGCATACTGTCC-3'(reverse); MMP-9 (h): 5'-TCCAGTACCAAGACAAAG-3' (forward) and 5'-TTGCACTGCACGGTTGAA-3' (reverse); RANK (h): 5'-CAAATGCAGACCCTGGA CCA-3' (forward) and 5'-AAACGCCAAAGATGATGGCA-3' (reverse); RANKL (h), 5'-CCTGTAT GCCAACATTTGCTTTC-3' (forward) and 5'-TTCCTCTCCAGACCGTAACTTAAA-3' (reverse); IL -6 (h): 5'-AGCAAAGAGGCACTGGCAGA-3' (forward) and 5'-GTACTCATCTGCACAGCTCTGG C-3' (reverse); TCF-4 (h): 5'-GCTCAGGGTATGGAACCGGC-3' (forward) and 5'-CCCTGTAGTC CTGGTGGCATG-3' (reverse); c-MYC (h): 5'-CCTGGTGCTCCATGAGGAGAC-3' (forward) and 5'-AGACTCTGACCTTTTGCCAGG-3' (reverse); and glyceraldehyde 3-phosphate dehydrogenase (GAPDH): 5'-TCAAGAAGGTGGTGAAGCAG-3' (forward) and 5'-AGTGGGAGTTGCTGTTGAAG T-3' (reverse). Values on the vertical axis represent 2(−ΔCt); ΔCt is the discrepancy between the target gene Ct and GAPDH Ct.
Western blot analysis
The cells were washed twice with phosphate-buffered saline (PBS; pH 7.4) and total proteins were extracted using radio immunoprecipitation assay buffer supplemented with 1% protease inhibitors, phosphatase inhibitors and phenylmethylsulfonyl fluoride (PMSF). The protein concentration was measured using the BCA Protein Assay Kit (Thermo PierceTM). The membrane was blocked with a solution containing 5% skim milk in TBS-T for 30 min and then washed in TBS-T. The membrane was incubated for overnight at 4°C with the following primary antibodies: E-cadherin (sc-7870, Santa Cruz Biotechnology), N-cadherin (ab76011, Abcam), β-catenin (#29822 94, Millipore), MMP-9 (#13667, Cell Signaling), IL-6 (#12153, Cell Signaling Technology), c-MYC (9E10, Santa Cruz Biotechnology), TCF-4 (#2565, Cell Signaling Technology), RANK (#4845, Cell Signaling Technology), P-ERK (#9101, Cell Signaling Technology), ERK (#9102, Cell Signaling Technology), GAPDH (#2118, Cell Signaling Technology), P-AKT (#9271, Cell Signaling Technology), AKT (#9272, Cell Signaling Technology), P-SRC (#2105, Cell Signaling Technology), SRC (#2108, Cell Signaling), GSK-3B (#9315, Cell Signaling Technology) and P-GSK-3B (#9336, Cell Signaling Technology). Horseradish peroxidase (HRP)-conjugated AffiniPure goat anti-rabbit IgG (H + L) and HRP-conjugated AffiniPure goat anti-mouse IgG (H + L) were obtained from Proteintech Group, Inc (Jackson) and used as secondary antibodies.
Cell migration
Cells were seeded in 6-well plates for the cell migration assay. After each treatment, a confluent monolayer was wounded using a 200 µL pipette tip. Images of wound closure were obtained under an inverted microscope after 48 h. The wound area was calculated using NIH ImageJ software.
Cell invasion assay
A total of 1 × 105 transfected cells were seeded into the top chamber of a 24-well polycarbonate Transwell chamber (8.0 µm pore size; Corning Incorporated, Glendale, AZ, USA) and then treated for 24 h with hRANKL or RANKL. The number of trypan blue-stained cells in five random fields was counted using an inverted microscope.
Luciferase reporter assay to assess Wnt/β-catenin activity
Cells were seeded into a 24-well plate 24 h prior to transient transfection with either 2 µg of TOPflash or FOPflash reporter plasmid along with 1 µg DNA using Lipofectamine 3000 (Thermo Fisher). The TOPflash luciferase reporter plasmid contains TCF-4-binding sites upstream of the luciferase gene, resulting in luciferase activity in the presence of active Wnt/β-catenin signalling. The FOPflash reporter plasmid, on the other hand, carried mutated TCF-4-binding sites. Total cell extracts were assayed for luciferase activity according to the manufacturer’s instructions (Promega).
Immunoprecipitation
Cells were lysed in lysis buffer (20 mM Tris-HCl pH 7.6–8.0, 100 mM sodium chloride NaCl, 300 mM sucrose, 3 mM MgCl2 [buffer A]; and 20 mM Tris pH 8.0, 100 mM NaCl, 2 mM ethylenediaminetetraacetic acid [buffer B]). Whole cell lysates obtained by centrifugation were incubated with antibodies specific for active β-catenin (Millipore) and TCF-4 (Cell Signaling Technology) (dilution 1:100) and protein A Sepharose beads (Amersham Biosciences) for 2 h at room temperature. The immune complexes were washed three times using wash buffer and examined by western blotting.
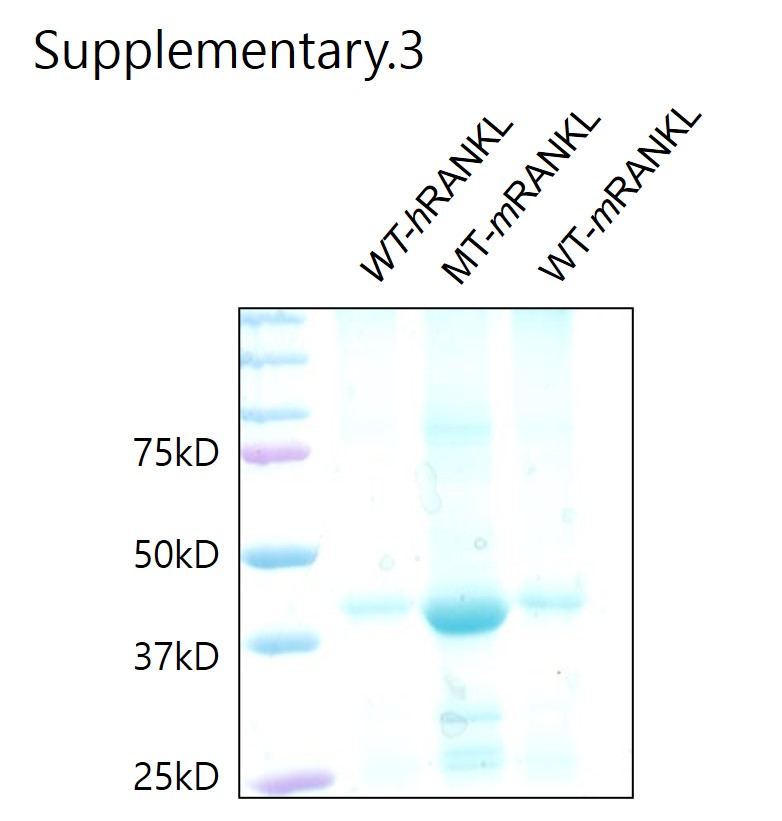
Site-directed mutagenesis and production and purification of mRANKL-MT
The RNA used for the cloning of RANKL cDNA was extracted from MC3T3-E1 cells (Korean Cell Line Bank, Seoul, Korea) expressing RANKL. The quality of the extracted RNA was verified by agarose gel electrophoresis and cDNA was prepared using the AccuPower RT PreMix Kit (Bioneer, Daejeon, Korea), according to the manufacturer’s instructions. Amplification and cloning of the RANKL fragment were carried out in a reaction mixture comprising KOD polymerase buffer, 10 mM dNTPs, 25 mM MgCl2, 10 μM primers (mRANKL-NdeI: 5'-CATATGAAGCCTGAGGCCCAGCC ATTTGC-3'; mRANKL-XhoI: 5'-CTCGAGGTCTATGTCCTGAACTTTGAAAGCC-3'; mRANKL (K180R)-F: 5'-CCCATCGGGTTCCCATCGAGTCACTCTGTCCTCTTG-3'; mRANKL (K180R)-R: 5'-CAAGAGGACAGAGTGACTCGATGGGAACCCGATGGG-3'; mRANKL (D189I, R190K)-F: 5'-CTCTTGGTACCACATCAAGGGCTGGGCCAAGAT-3'; mRANKL (D189I, R190K)-R: 5'-ATC TTGGCCCAGCCCTTGATGTGGTACCAAGAG-3'; mRANKL-MT (H223F, H224Y)-F: 5'-AA CA TTTGCTTTCGGTTTTATGAAACATCGGGAAGCG-3'; or mRANKL-MT (H223F, H224Y)-R: 5'-CGCTTCCCGATGTTTCATAAAACCGAAAGCAAATGTT-3'), 2.5 U of KOD DNA polymerase (EMD Millipore, Billerica, MA, USA) and 2 μL of RANKL gene construct as the template.
The thermal cycling conditions were as follows: initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s and extension at 70°C for 30 s. The PCR product obtained was cloned into the NdeI/XhoI site of the GST-30a vector (Novagen, Madison, WI, USA). Mutations at positions 180, 189–190 and 223–224 were introduced using megaprimers [26]. The PCR product was transformed into Escherichia coli BL21-CodonPlus (DE3)-RIPL (Novagen) by electroporation (5 ms, 12.5 kV/cm) and the transformed cells were cultivated in Luria-Bertani broth containing kanamycin (50 μg/mL, T&I, Daejeon, Korea). Plasmids were purified using the QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA, USA). The cloned product was confirmed by sequencing. All sequence analyses were carried out using programs in Vector NTI Advance 9.1.0 (Invitrogen, Carlsbad, CA, USA). The recombinant plasmid carrying mRANKL-MT was expressed from a single E. coli BL21-CodonPlus (DE3)-RIPL colony using previously described methods [26].
Purification of mRANKL-MT
E. coli cells expressing mRANKL-MT were cultivated in 1 L of an auto-induction medium supplemented with kanamycin (50 μg/mL), as previously described. After centrifugation at 6000 ×g for 20 min at 4°C, the pelleted cells were resuspended in 10 mL of lysis buffer (20 mM sodium phosphate, 500 mM NaCl, 10 mM imidazole, pH 7.4) supplemented with 0.1 mg/mL lysozyme and 0.1 mM PMSF.
Glycerol (20% v/v; CARLO ERBA, France) was added to the cell suspension and the cells were sonicated and centrifuged at 15,000 ×g for 10 min at 4°C. The supernatants were passed through 0.2 μm paper filters and applied to Ni2+-affinity chromatography HisTrap FF columns (1 mL; GE Healthcare Life Science, Piscataway, NJ, USA) equilibrated with binding buffer (20 mM sodium phosphate, 500 mM NaCl, 10 mM imidazole, 5 mM dithiothreitol, pH 7.4). The columns were subsequently washed using binding buffer supplemented with 20 mM imidazole.
After washing, bound protein was eluted using elution buffer (Qiagen). The eluted protein was dialysed against a dialysis buffer (20% v/v glycerol in PBS) in a 10,000 MW Slide-A-Lyzer Dialysis cassette (Thermo Fisher Scientific, Waltham, MA, USA). The purified protein was vacuum concentrated (Savant Instruments, Holbrook, NY, USA) and analysed by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). Protein concentrations were calculated using the Bradford assay. For endotoxin removal, an additional washing step was introduced after the initial wash for chromatography.
Animal study
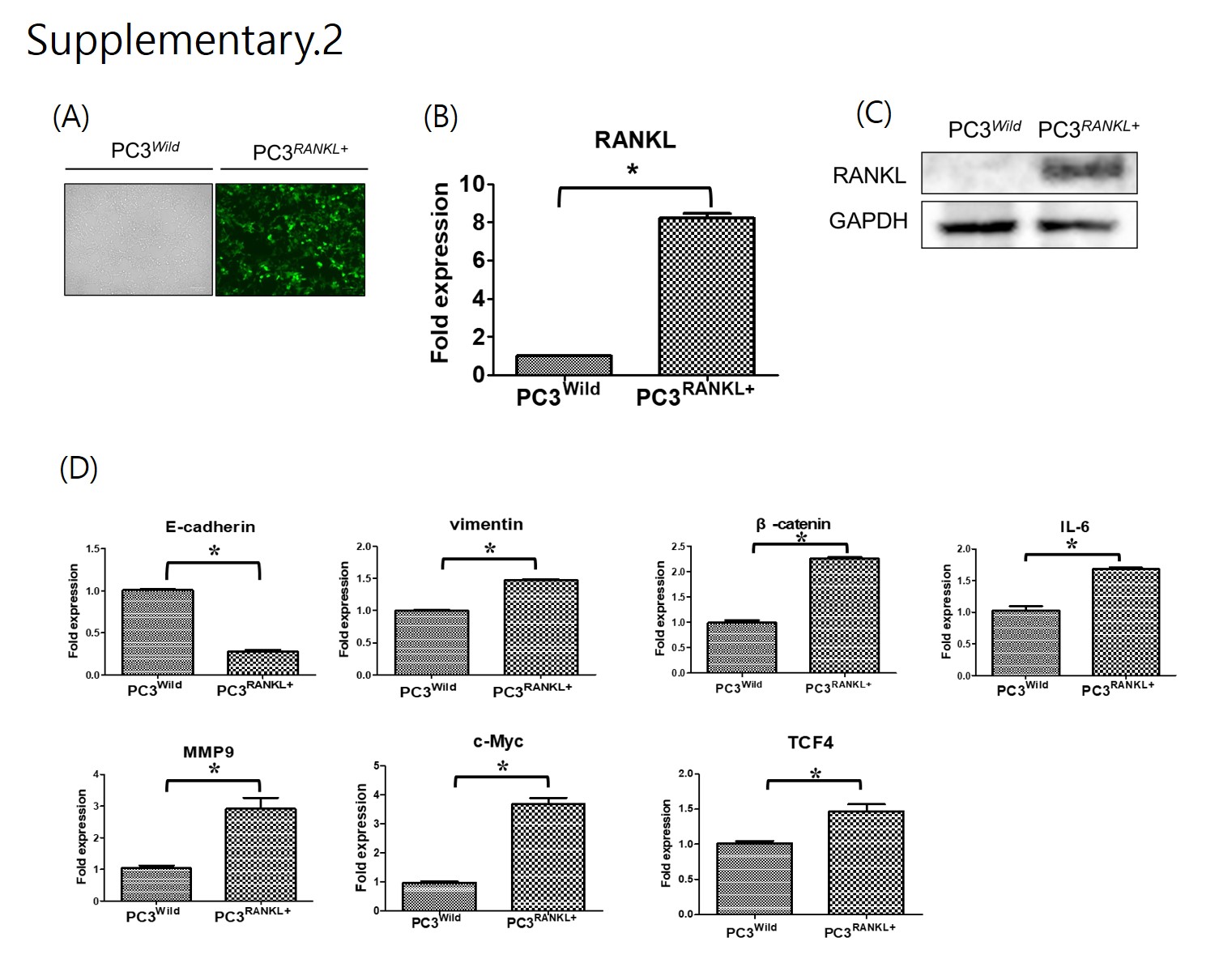
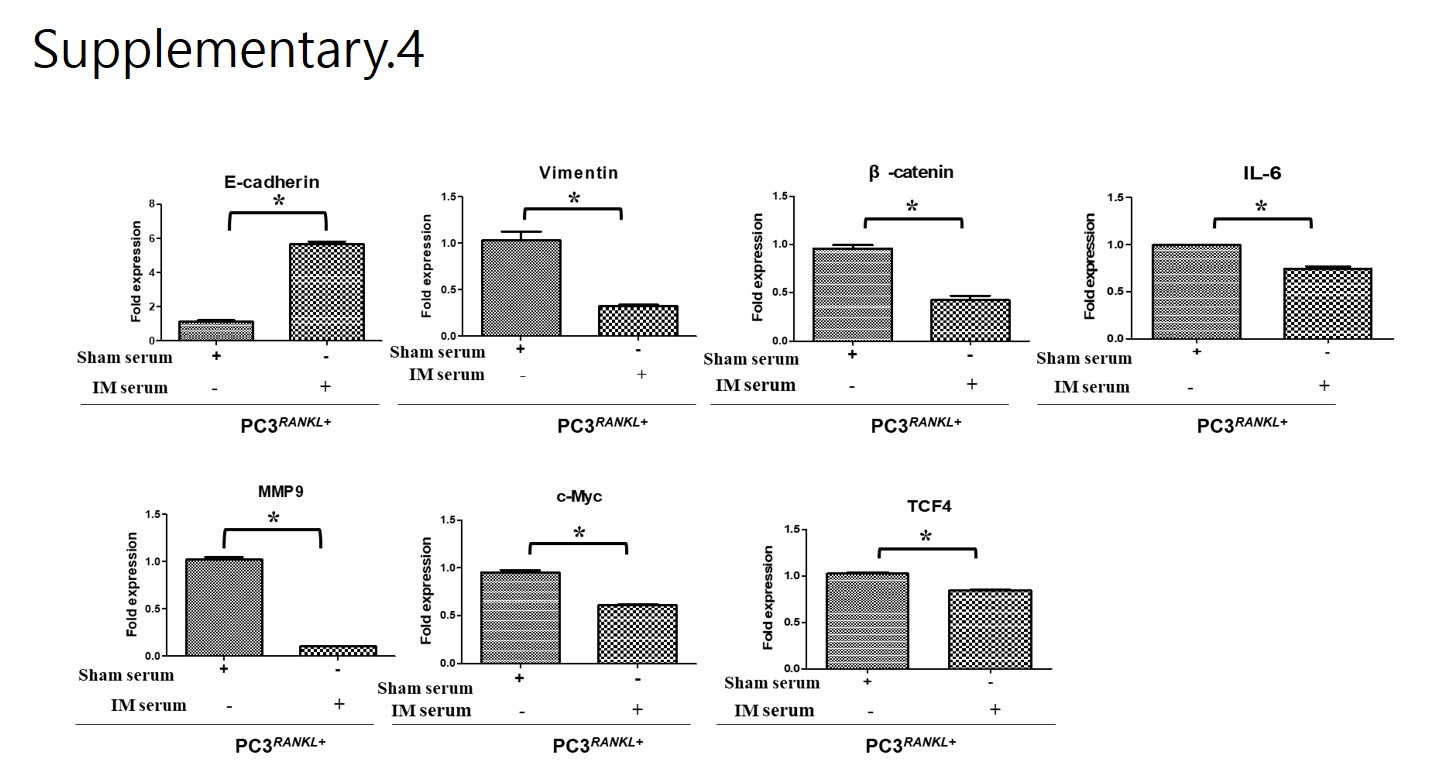
The animal experimental protocol was approved by the Institutional Animal Care and Use Committee, Chosun University, Gwangju, Korea (CIACUC2019-A0015). All experiments were performed in accordance with relevant guidelines and regulations. Five-week-old male athymic nude mice (BALB-c/nu, Orient Bio Co. LTD, Seoul, Korea) were used to generate a xenograft model by intracardiac injection of PC3Wild, PC3+RANKL (RANKL overexpression), or PC3+RANKL + IM (immunisation) cells. Following immunisation, mice were divided into an immunisation group and a non-immunisation group. The Sham group was immunised by a subcutaneous injection of PBS, while the immunisation group was injected subcutaneously with mRANKL-MT (100 µg/kg three times every 2 weeks). Mouse sera and tissue samples were collected according to indicated schedule.
In vivo bioluminescence measurement
Tumour-bearing tissues were subjected to in vivo bioluminescence imaging using a Living Image® 4.5.4 IVIS Imaging System (Perkin Elmer). For luciferase imaging, D-luciferin (Promega) was injected intraperitoneally before imaging. Quantitative detection of luciferase was performed as follows: regions of interest (ROIs) were drawn to capture detected fluorescence, and auto-regions ROIs were used to precisely outline the target region.
Quantitative analysis of RANKL
The amount of RANKL in mouse serum was measured using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, USA) according to the manufacturer’s protocol. Absorbance was measured in a colorimetric microplate reader (BioTek, USA) at 450 nm.
Measurement of anti-RANKL antibody titers
Serum samples obtained from immunised mice were serially diluted with PBS containing 0.02% sodium azide and 2% bovine serum albumin (BSA), and then applied to ELISA plates (Sigma-Aldrich) coated with mouse recombinant tumour necrosis factor ligand superfamily member 11 (TNFSF11; 10 μg/mL, R&D Systems). Reactivity of serum antibodies to the target protein was determined using an HRP-conjugated goat anti-mouse IgG secondary antibody (Thermo Fisher Scientific) at a dilution of 1/1000 in PBS/0.02% sodium azide/2% BSA. After development with 1,2- phenylenediamine dihydrochloride (0.4 mg/mL in 0.066 M disodium phosphate, 0.035 M citric acid and 0.01% hydrogen peroxide), absorbance was measured in an ELISA plate reader at 450 nm.
Statistical analysis
Data are expressed as the mean ± standard deviation (SD) from three independent experiments. GraphPad Prism version 6.0 software for windows was used to analyse in vitro and in vivo data. Statistical significance for pairwise comparison was evaluated using an unpaired t-test or one-way analysis of variance (ANOVA) with Turkey’s post-hoc test. Results were considered significant at *p < 0.05.
{kind=link}
{kind=link}
{kind=link}
{kind=link}