Identification of de novo variant in ASD probands
We analyzed a sample set consisting of 168 ASD probands and 326 parents from 163 pedigrees recruited from Department of the Child and Adolescent Psychiatry, Shanghai Mental Health Center. Among the cohort, there are 5 multiplex family containing two ASD children and the rest 158 family are trios having one ASD child. The fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) were used for ASD diagnoses by trained psychiatrists.
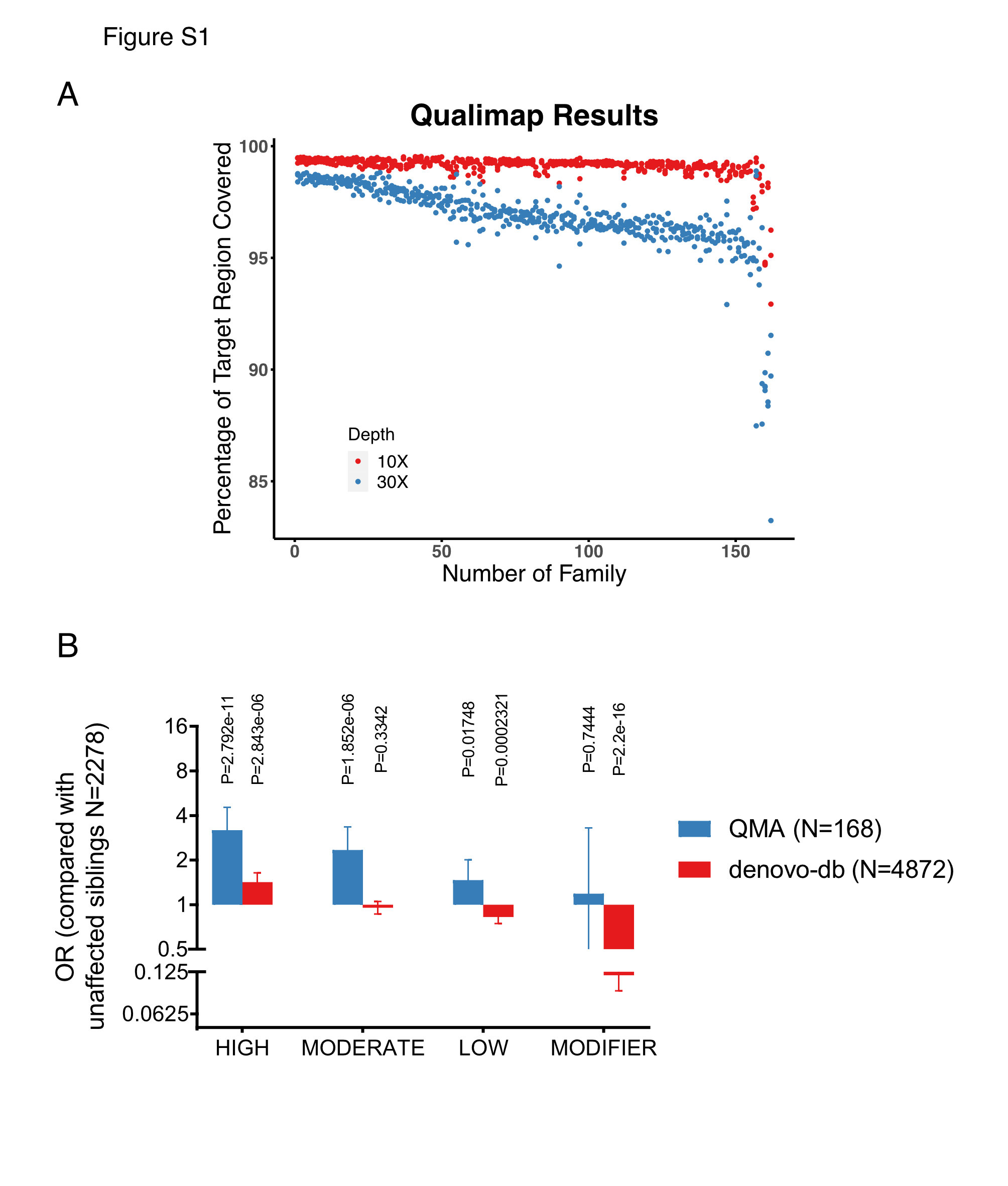
Proportion of the target exome region covered with ≥ 10x or 30x of reads indicates sufficient coverage (Figure. S1A). After performing the multidimensional scaling of the genotyping data of common exonic SNPs was performed by using PLINK (a whole genome association analysis toolset)(9), we found that all probands in this cohort were included in the cluster of East Asian individuals (Figure. 1A).
After performing variant filtering, we discovered a set of 442 de novo mutations (DNMs) (Table S1). We classified SNV and INDELs into three classes, including HIGH-impact. MODERATE-impact, and Possible damaging. The HIGH- and MODERATE-impact were defined by VEP (Ensembl Variant Effect Predictor, https://asia.ensembl.org/info/docs/tools/vep/index.html). Briefly, the HIGH-impact variants usually lead to truncation of protein product, such as gain or loss of STOP codons as well as frameshift-causing INDELs. We identified 11 HIGH-impact SNVs and 8 HIGH-impact INDELs (Figure. 1B, C). Interestingly, among the 11 genes containing HIGH-impact SNVs, there are 5 genes previously reported in the SFARI gene list (SCN2A, POGZ, MECP2, SRCAP, TCF4). However, in the 7 genes containing HIGH-impact INDELS (there are 2 recurrent INDELs in SYNGAP1 genes), only SYNGAP1 and CUX1 are reported in the SFARI gene list, suggesting that there are substantially non-SFARI ASD gene in the Chinese cohorts (Figure. 1B, C).
MODERATE-impact variants were defined as protein sequence changing, but not truncating, such as missense variants and inframe INDELs. We found there are 15 inframe INDELs classified as MODERATE-impact variants (Table S1). To further categorize the severity of missense variants, we annotated missense into a new class, named Possible damaging missense DNMs, which were defined as the variants predicted to be damaging by at least two of the seven following prediction algorithms: SIFT(10), PolyPhen-2 HumVar(11), PolyPhen-2 HumDiv(11), LRT(12), Mutation Taster(13), Mutation Assessor(14) and PROVEAN(15) annotated by dbNSFP4.0a(16, 17). We found 64 Possible damaging missense DNMs (Table S1).
Next we statistically assessed the observed number of de novo variants in each gene using Transmission and De Novo Association Test-Denovo (TADA-Denovo) and identified one gene significantly enriched for de novo mutations (SYNGAP1 q val < 0.05) (Table S2). Overall, de novo ASD risk genes detected in ASD probands from the Chinese cohort showed little overlapped with the list of de novo ASD risk genes in ASD probands from the Japanese cohort(Figure. 1D)(5). Only a few SFARI genes, including SYNGAP1, POGZ and NCOA6 were found in both East Asian cohorts.
Odds Ratio may not be a good measure of genetic risks for ASD.
We next re-annotated DNM data from 4872 ASD probands and 1943 unaffected siblings originally from db-denovo v.1.6.1 with the same pipeline as used for our dataset. By comparing the proportion of individuals carrying one or more HIGH, MODERATE, LOW and MODERATE impact mutations in the case groups with controls, We confirmed that carriers of HIGH impact DNM were significantly enriched in both our cohort and the db-denovo ASD cohort (p= 2.792 × 10-11, odds ratio [OR] = 3.182105 in our ASD cohort; p= 2.843 × 10-6, odds ratio [OR] = 1.418789 in the db-denovo ASD cohort, Figure. S1B). However, there was no enrichment of MODERATE-impact DNM carriers with lower ORs in db-denovo case cohorts (p= 1.852 × 10-6, odds ratio [OR] = 2.343237 in our ASD cohort; p= 0.3342, odds ratio [OR] = 0.9531462 in the db-denovo ASD cohort, Figure. S1B). And there was a statistically significant enrichment of LOW impact DNM carriers with a pair of contradictory ORs in both case cohorts (p= 0.01748, odds ratio [OR] = 1.464373 in our ASD cohort; p= 2.321 × 10-4, odds ratio [OR] = 0.8257816 in the db-denovo ASD cohort, Figure. S1B).
Furthermore, the odds ratio of MODIFIER impact DNM carriers in the db-denovo ASD cohort suggests that a type of mild mutations inhibit the onset of ASD(p= 0.7444, odds ratio [OR] = 1.185185 in our ASD cohort; p= 2.2 × 10-16, odds ratio [OR] = 0.1182785 in the db-denovo ASD cohort, Figure. S1B). Taken together, these results indicate that odds ratio can only be partially used to determine the effect of different mutation types on the incidence of ASD.
Identification of CNVs in ASD risk genes with the WES dataset
Although the gold standard for copy number variations detection is the chromosomal microarray analysis (CMA), various toolkits has emerged to identify CNVs with the whole-exome sequencing (WES) dataset(18). However, the current reported algorithms for CNV detection is not optimal for the WES dataset and incompatible with the GRCh38/hg38 reference genome.
We applied a germline CNV calling protocol based on GATK cohort mode (version 4.1.4.1) (See Supplementary Methods) and identified numerous de novo CNVs in the probands (Table S3). To exclude the false positive hits, we set 2 standards for CNV screening. First, selection of duplication or deletion signals appearing in more than 2 continuous exons. Second, CNVs should fulfill the HIGH-impact criterial, leading to protein truncation, such as deletion of START or STOP codons.
To prioritize ASD risk genes, we first examine CNVs happened in the known SFARI genes (Figure. 2A-G). We found 8 CNVs exhibiting duplication or deletions in known SFARI genes, such as duplications of AMT, RAI1, TBC1D23, and deletions of TBR1, SHANK3, MECP2, GIGYF1 (Figure. 2A-G). We further validated the CNV results by performing quantitative PCR (Figure. 2H), confirming the feasibility and faithfulness of our new methods.
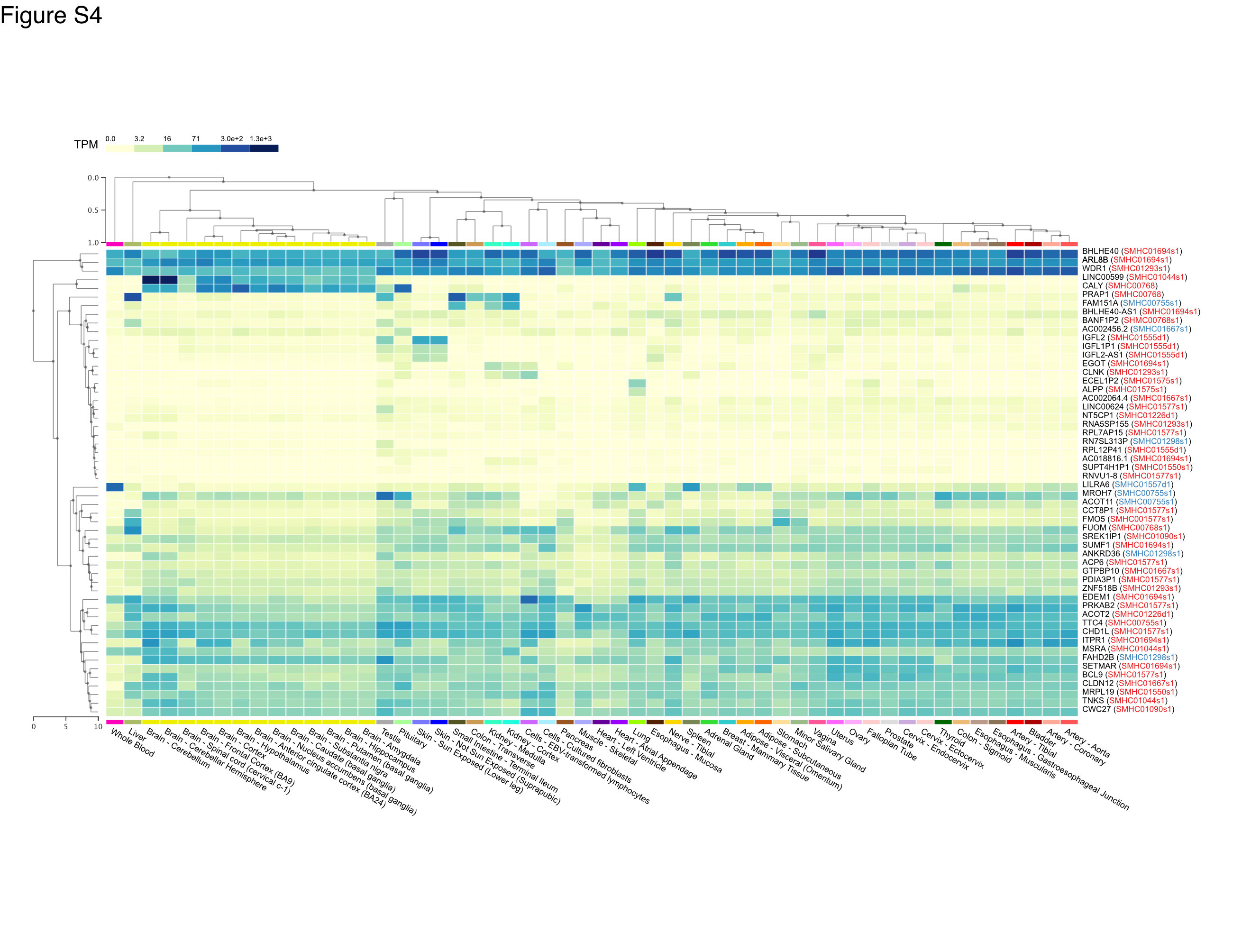
Importantly, we further identified de novo large CNVs, containing multiple genes (Figure. S2A-H, Figure. S3A-H). To investigate whether these candidate genes may be involved in brain development, we examine the expression pattern of candidiate genes which exhibited either duplications or deletions in ASD patients in the GTEx Analysis Release V8 database (dbGaP Accession phs000424.v8.p2). We found that numerous candidate genes indeed were expressed in the central nervous system (Figure. S4), suggesting that genes implicated in these de novo large CNVs may contribute to pathogenesis of ASD.
{kind=link}
{kind=link}
{kind=link}
{kind=link}