Experimental animals
Experiments were conducted on C57BL/6 mice (male, 19 to 22 g), which were purchased from the Experimental Animal Center of Chongqing Medical University (Chongqing, China). Prior to the experiments, standard chow and water were given to the mice ad libitum. All mice were housed in cages in a room with a controlled temperature of 23℃ and humidity of 60% under a 12-h light/12-h dark cycle. The animal experiments were approved by the China Association of Laboratory Animal Care, and all efforts were made to minimize suffering. No mice were sacrificed unexpectedly during any experiments.
Adenovirus
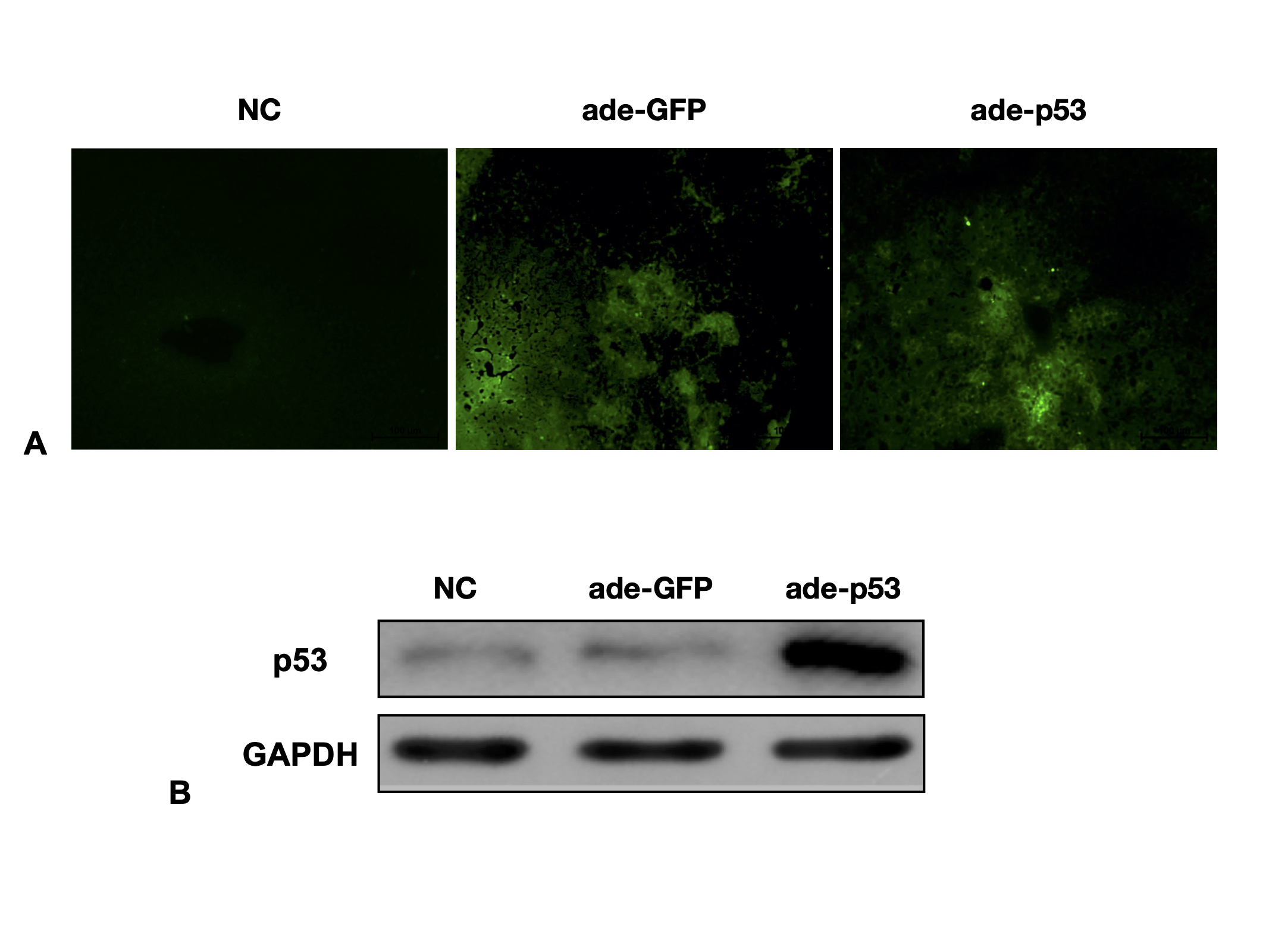
Adenovirus (Ade) expressing green fluorescent protein (GFP) and a sequence targeting P53 was constructed by ABM (Nanjing, China). Ade-GFP was used as a control. Five days before the surgeries, mice were administered 5 × 1011 genome-equivalents of adenovirus by tail vein injection.
Adeno-associated virus-8
Adeno-associated virus-8 (AAV8) with green fluorescent protein (GFP) and a sequence targeting miR-34a was constructed by ABM (Nanjing, China), as previously described. AAV8-GFP was used as a control. Two weeks before the surgeries, mice were administered 5 × 1011 genome-equivalents of AAV8 by tail vein injection.
Experimental groups and PH
Mice were randomly divided into groups as follows:
Overexpression of P53
(1) Normal control group (NC): mice underwent sham or PH surgery (sham, n = 9 per time point; PH, n = 9 per time point); (2) Ade-GFP group: 5 days after tail vein injection of Ade-GFP, mice underwent sham or PH surgery (sham, n = 9 per time point; PH, n = 9 per time point); (3) Ade-p53 group: 5 days after tail vein injection of Ade-p53, mice underwent sham or PH surgery (sham, n = 9 per time point; PH, n = 9 per time point).
Overexpression of miR-34a
(1) Normal control group (NC): mice underwent sham or PH surgery (sham, n = 9 per time point; PH, n = 9 per time point); (2) GFP group: 2 weeks after tail vein injection of AAV8-GFP, mice underwent sham or PH surgery (sham, n = 9 per time point; PH, n = 9 per time point); (3) AAV-anti-miR-34a group: 2 weeks after tail vein injection of AAV-anti-miR-34a, mice underwent sham or PH surgery (sham, n = 9 per time point; PH, n = 9 per time point).
MCA experiment
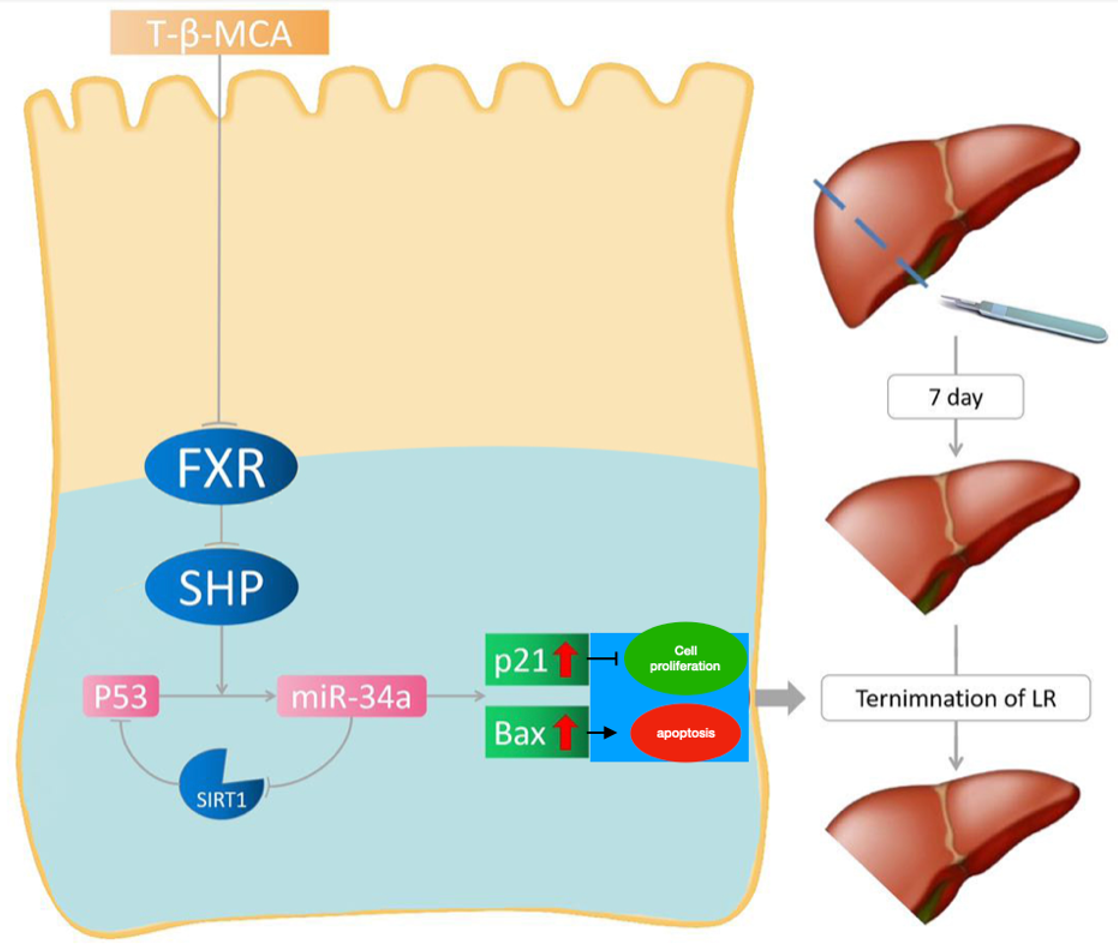
(1) Normal diet group (ND): mice were fed a normal diet and underwent sham or PH surgery (sham, n = 9 per time point; PH, n = 9 per time point);
(2) T-β-MCA group: mice were fed normal diet and gavage with extra T-β-MCA (400 mg/kg, Steraloids, Cat# C1899-000) 1 day before PH and every 3 days after PH. Mice underwent sham or PH surgery (sham, n = 9 per time point; PH, n = 9 per time point).
Mice underwent classical PH as previously described[12]. Briefly, mice were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg). Then, the left and median lobes of the liver were ligated and resected. The same surgical procedure was performed on the sham groups without conducting PH. Animals were euthanized after administration of an inhalable anesthetic (2% to 3% isoflurane) at designated times (0, 2, 3, 5, 7, 10, 14 days) after operation, and whole blood was collected via portal vein puncture. Liver tissues were quickly perfused with ice-cold phosphate-buffered saline (1×) before they were excised and weighed. The liver/body weight ratio was calculated using the following equation: liver/body weight ratio = (remnant liver weight [g]/body weight [g]) × 100%.
Cell isolation and purification
Primary hepatocytes were isolated according to the collagenase perfusion method as previously described[13]. Briefly, mice were fully anesthetized and decontaminated, and the liver was immediately infused with buffers 1 and 2 before removal and transfer to a 100 mm cell culture plate for mechanical dissociation. The suspension (1 mL; 5 × 106 hepatocytes/mL) was dispensed into each well of 12-well culture plates and incubated at 37°C in an atmosphere of 5% CO2 for later use. The viability and purity of hepatocytes were assessed by light microscopy to ensure the presence of at least 90% hepatocytes in the suspension.
Cell transfection
Primary mouse hepatocytes were transfected with adenovirus particles with knock-in of wild-type P53 according to the manufacturer’s instructions. For the control, the cells were transfected with NC or GFP in a manner identical to that used for transfection of P53. All primers were synthetized by ABM (Nanjing, China). Subsequently, the transfection efficiency was confirmed by fluorescence detection and western blot analysis. The cells were collected for further study.
Hep-3B, Huh7 and HepG2 cells (3×105) were seeded in each well of 6-well plates and cultured for 24h at 37 °C. The cells were then coincubated with 10 μl Lipofectamine® 2000 (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 4 μg plasmid pRFP-p53α or pRFP at 37 °C for 6 h. Subsequently, the cells were treated with fresh medium followed by an additional 24 h of incubation at 37 °C. Subsequently, the transfection efficiency was confirmed by fluorescence detection and western blot analysis. The cells were collected for further study.
Measurement of bile acid metabolites
Liver tissue was extracted from PH mice at the indicated times. Then, the samples were first suspended in sodium hydroxide (1 M) and then resuspended in methanol and pyridine. Thereafter, MCF was added and mixed by vortexing to initiate derivatization; chloroform and bicarbonate (50 mM) were then added sequentially and mixed; finally, the organic layers of samples were extracted by centrifugation (12000×g, 5 min, 24°C). The derivatized samples were transferred into glass inserts inside liquid chromatography (LC) vials after adding anhydrous sodium sulfate, and empty tubes subjected to the same process were used as negative controls. The derivatized extracts were analyzed with an ultrahigh-performance liquid chromatography (UHPLC-MS/MS) system (Agilent, 1290-6460) to quantify the differential expression of metabolites[14].
Liver histopathology
Liver tissues were fixed in 10% neutral formalin, embedded in paraffin, and then cut into 5 μm thick sections. The sections were baked at 60°C for 1 h and dehydrated with gradient ethanol and xylene. Then, the sections were stained with eosin for 30 min. After rinsing with running water for a few seconds, the sections were dehydrated with gradient ethanol and xylene and sealed with neutral gum. Histopathological changes of the liver were observed under a microscope.
Detection of liver function
Blood samples were collected from mice at the indicated time points and then centrifuged at 3000 r/min for 5 min to collect the upper translucent serum layer. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured at the indicated times using a standard automatic biochemistry analyzer in the clinical biochemical laboratory.
Immunohistochemical evaluation
Liver tissues were fixed in 10% neutral formalin, embedded in paraffin, and then cut into 5 μm thick sections. The sections were dehydrated with gradient ethanol and xylene and subjected to heat-induced antigen retrieval using citrate. Then, the sections were permeabilized with 0.3% Triton for 15 min. After eliminating endogenous peroxidase activity with 3% hydrogen peroxide for 15 min, the sections were blocked with 10% fetal sheep serum for 30 min. Then, the sections were incubated with primary antibodies (1:50 dilution) overnight at 4°C. After rinsing with PBS 3 times, the sections were incubated with species-matched secondary antibodies (1:200 dilution) for 1 h at room temperature and treated with 3,3'-diaminobenzidine for 5 min at room temperature. After washing, the sections were stained with hematoxylin for 30 sec at room temperature and washed with flowing water for a few seconds. Following dehydration, the sections were sealed with neutral resin, and specific staining was visualized by light microscopy as described previously[15]. The following primary antibodies were used: anti-P53 (cat. No. Ab26; 1:50; Abcam Inc.) and anti-PCNA (cat. No. Ab92552; 1:50; Abcam Inc.).
RNA isolation and quantification
Total RNA was extracted from liver tissues using a TRIzol kit (Takara, Otsu, Japan) and was reverse transcribed into cDNA using a PrimeScript™ 1st Strand cDNA synthesis kit (Takara, Otsu, Japan). The primer sequences of miR-34a and U6 were designed and synthesized by Guangzhou RiboBio Co., LTD (Guangzhou, China). Next, qPCR assays were performed using miDETECT A Track miRNA qRT-PCR Starter Kit(RiboBio Co., LTD) and a cDNA template on an Applied Biosystems 7500 Real-time PCR system (Applied Biosystems; Thermo Fisher Scientific Inc). With U6 serving as the internal reference, the relative expression of genes was calculated using the 2-∆∆Ct method [16].
Western blot analysis
Total proteins were extracted and denatured for 10 min at a temperature of 100°C. A total of 40 µg of protein was loaded per lane. After electrophoresis by SDS/PAGE, proteins were electrotransferred onto polyvinylidene difluoride membranes. Then, the membranes were blocked for 1 h and incubated with primary antibodies at 4℃ overnight. Primary antibodies against the following antigens were used: P53 (cat. No. Ab26; 1:1000; Abcam Inc.), Ace-P53 (cat. No. 2570S; 1:1000; CST.), SIRT1 (cat. No. 8469S; 1:1000; CST), cleaved Caspase3 (cat. No. Ab231289; 1:1000; Abcam Inc.), P21 (cat. No. Ab188224; 1:1000; Abcam Inc.), Bax (cat. No. Ab32503; 1:1000; Abcam Inc.), NR1H4 (FXR) (cat. No. Ab187735; 1:1000; Abcam Inc.), and NR0B2 (SHP) (cat. No. Ab186874; 1:1000; Abcam Inc.). The membranes were then incubated with species-matched secondary antibodies. Protein bands were visualized using the Bio-Rad ChemiDoc XRS system (Hercules, CA). All images were analyzed using NIH ImageJ software.
Ethynyl-2′-deoxyuridine cell proliferation assays
Cell proliferation was assessed by the incorporation of 5-ethynyl-2′-deoxyuridine (BeyoClick™ EdU-488 In Vitro Imaging Kit, Cat. No. C0071S, Beyotime Biotechnology Inc.) into DNA according to the manufacturer’s instructions. Briefly, hepatocytes were isolated and cultured as previously described. EdU was added to the culture at a concentration of 100 nmol/L. According to the standard formaldehyde fixation protocol, the cells were permeabilized, fixed and incubated with the reaction cocktail for 30 min. The images of stained cells were captured by fluorescence microscopy[17].
TUNEL apoptosis assays
The TUNEL reaction was performed using the one step TUNEL apoptosis assay kit-green fluorescein (Cat. No. C1086, Beyotime Biotechnology Inc.) according to the manufacturer's instructions. Briefly, liver sections were deparaffinized and dehydrated. The sections were incubated in immunostaining wash buffer (0.1% Triton X-100 in PBS) for 5 min, labeled with 50 μl of TUNEL reaction mixture and incubated at 37°C for 1 h in the dark, followed by counterstaining with DAPI. After washing, slides were mounted and observed under an immunofluorescence microscope.
Statistical analysis
Statistical analyses were conducted using SPSS 22.0 software, and all values are expressed as the mean±SD. Statistical significance of differences was calculated using t-tests for parametric data involving two groups and one-way analysis of variance with Tukey’s test for multiple groups. Comparisons among datasets at different time points were analyzed by repeated measurement ANOVA. The log-rank test was used to assess the differences between the survival curves. Differences were considered statistically significant if P<0.05.
{kind=link}
{kind=link}