Characterizations of the NIR-CDs.
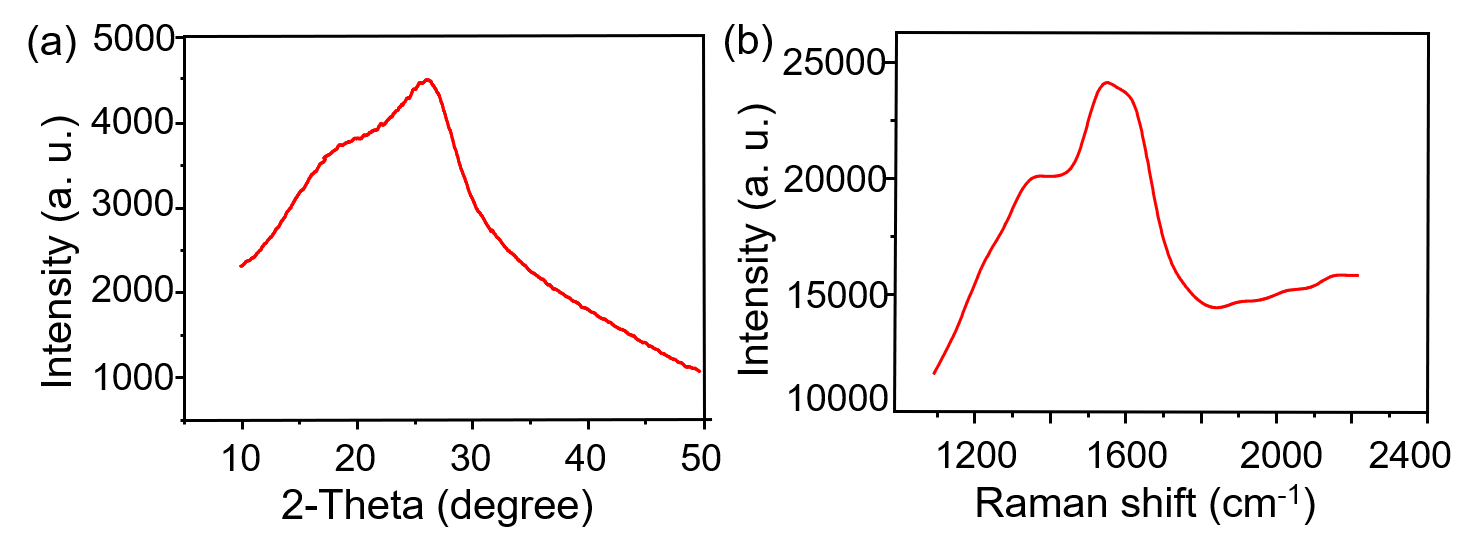
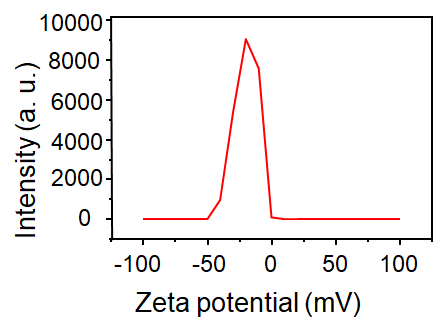
The NIR-CDs were prepared with a facile microwave-assisted carbonization method using GSH and formamide as the raw materials and solvent, respectively. To characterize the morphology, size, chemical composition and surface state of the harvested NIR-CDs, a series of measurements e.g. TEM, XRD, FT-IR, XPS, Raman and Zeta potential were severally performed. As seen from the TEM image (Fig. 1A), the NIR-CDs present very good mono-dispersity, uniform and spherical morphologies, and a narrow particle distribution with a mean size of 3.9 nm (Fig. 1B). Nonetheless, there are none lattice fringes are observed in the HR-TEM image (Fig. 1C), indicating that the NIR-CDs are mostly noncrystalline. In the XRD pattern (Additional files Fig. S1a), a typical peak at 26° is assigned to the (002) plane of graphite, which further verifies the noncrystalline graphite structure of the NIR-CDs [23]. Furthermore, the graphite structure of NIR-CDs also confirmed by the Raman spectrum (Additional files Fig. S1b). As shown therein, two distinct peaks centered at 1550 and 1342 cm−1 represent the typical G-band and D-band, respectively. Simultaneously, a low ratio of D to G clearly authenticates the dominate pristine carbon in the NIR-CDs [24]. In the FT-IR spectrum of NIR-CDs (Fig. 1D), a broad and strong absorption band range from 3100 to 3500 cm−1 is attributed to the stretching vibrations of O–H and N–H, implying the abundant existence of hydrophilic hydroxyl and amino groups. Typical stretching vibrations absorption peaks of C=O and C=C/C=N are easily observed at 1689 and 1583 cm−1, respectively. The peaks at 1160, 1249 and 1390 cm−1 severally belong to the stretching vibrations of C-C, C–O and C–N. The absorption band (1000–1100 cm−1) is assigned to C=S and oxidized S bonds [25]. These FT-IR assignments are further clearly confirmed by XPS analysis (Fig. 1E). Representative peaks of C 1s, N 1s, O 1s, and S 2p are observed at 283, 398, 529, 161 eV, respectively [26], indicating that the NIR-CDs mainly contain C, N, O and S elements (atom ratio, C:O:N:S = 63.9:16.3:19.4:0.4). High resolution C 1s spectrum (Additional files Fig. S2a) reveals four typical peaks at 284.8, 286.3, 288.0 and 290.1, which is attributed to C=C/C−C, C−N/C−O, C=N/C=O and N−C=O, respectively [27]. Three obvious peaks of pyridine-like N, graphitic N, and pyrrole-like N at 398.5, 400.0, and 402.6 eV are severally observed in high resolution N 1s spectrum (Additional files Fig. S2b) [28]. O 1s XPS spectrum shows two unique peaks of C−OH and C=O at 531.3 and 533.6 eV [29], respectively (Additional files Fig. S2c). Besides, in the high solution S 2p spectrum, four binding energies at 162.2, 163.4, 164.8 and 169.7 were discriminated, corresponding to thiolate, 2p3/2 and 2p1/2 of thiophene S, and oxidized S, respectively [30]. Finally, zeta potential measurement reveals that the NIR-CDs are negatively charged (ζ = − 19.2 mV, Additional files Fig. S3), which brings strong electrostatic exclusion and good colloid stability.
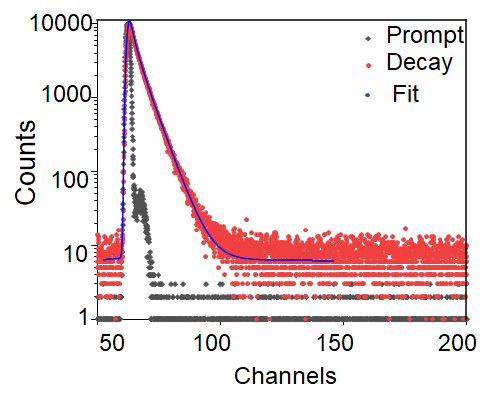
Thereafter, the photophysical properties including Uv-vis absorption, fluorescence spectrum and fluorescence lifetime of the NIR-CDs were inspected, respectively. As shown in Fig. 2A, three distinct absorption bands i.e., 240–300 nm, 350–450 nm, and 550–750 nm are observed, which are generally ascribed to the typical π → π* transition of the aromatic C=C bond, π → π* and n → π* transitions of the aromatic π system containing C=O, C=N, and C=S bonds [25, 31, 32], respectively. In Fig. 2b, the NIR-CDs reveal strongly deep-red emission from 625 to 725 nm with a sharp peak centered at 680 nm under different excitation wavelengths. Such an excitation-independent fluorescence emission property is usually attributed to the surface states/defects induced emission [33]. In addition, the average fluorescence lifetime of the NIR-CDs is measured and calculated to be 3.2 ns with bi-exponential decays (Additional files Fig. S4), and the absolute fluorescence quantum yield is measured to be 18.4% under an optimal excitation of 420 nm. In our previous reports [21, 23], the NIR-CDs have been demonstrated excellent tolerance to photobleaching under the excitation of ultraviolet light, good storage stability at ambient environment and competitive agent in light-harvesting and electron transfer from photosystem II (PS II) to photosystem I (PS I) in chloroplasts [18, 34].
NIR-CDs treatment significantly improved growth and photosynthesis of T. hemsleyanum
Figure 3A is the phenotypic characteristic of T. hemsleyanum exposed to 0.05 mg/mL NIR-CDs on day 30. The treatment enables T. hemsleyanum to grow more exuberantly than the control group. As shown in Fig. 3B, compared to the control, root length, stem length, leaf area, net photosynthetic rate, stomatal conductance, transpiration rate, chlorophyll fluorescence parameter Fv/Fn and chlorophyll content were significantly increased by 38.36± 6.6%, 41.40 ± 1.9%, 93.84 ± 9.2%, 16.25± 1.2%,122.51± 10.6%, 120.14 ± 11.1%, 80.36 ±7.1% and 87.34± 3.2%, respectively. Intercellular CO2 was decreased by 51.93± 3.8% than the control.
Confocal images were captured using Laser-scanning confocal fluorescence microscope after incubation with 0.05 mg/mL NIR-CDs. As shown in Fig. 3A, the red fluorescence signals from NIR-CDs observed under 514 nm excitation were widely distributed in root, stem, and leaf of T. hemsleyanum, which confirmed that NIR-CDs could penetrate cell wall into vascular bundle system, and were transported to the aerial parts.
Metabolome profiling and KEGG enrichment analysis of differential metabolites
We profiled the widely-targeted LC-MS/MS based metabolome of the samples from the control (denoted as CK) and the NIR-CDs-treatment groups (denoted as TH), which represented the impact of the NIR-CDs on metabolite accumulation of T. hemsleyanum. We detected 493 secondary metabolites grouped into 8 classes (Additional files Table S1), including 182 flavonoids, 167 phenolic acids, 20 lignans and coumarins, 64 alkaloids, 15 terpenoids, 26 tannins and others. The differentially accumulated metabolites (DAM) between the control and the experimental groups (CK_vs_TH) were screened using the variable importance in projection (VIP)≥ 1 from the OPLS-DA model and fold change ≥ 1.5 (upregulated) or ≤ 0.667 (downregulated). A total of 191 DAM were identified in CK_vs_TH (Additional files Table S2). These secondary metabolites can be mainly categorized into the classes of flavonoids and phenolic acids, including 106 flavonoids, 43 phenolic acids, 12 lignans and coumarins, 9 alkaloids, and others. Overall, flavonoids were more inclined to accumulate in CK than in TH. The content of phenolic acids, lignans and coumarins were significantly improved by CDs-treatment (Fig. 4A).
We focused on the two classes of secondary metabolites (flavonoids and phenolic acids) likely to be major contributors to biological activity (Fig. 4B). Flavonols and flavonoid carbonoside were identified with a series of glycoside derivatives of kaempferol, quercetin, and apigenin, which made up the majority of the DAMs detected for CK _vs_TH compared samples. Based on fold changes and VIP values, 77 out of 106 flavonoids were identified as down-accumulated significantly by CDs-treatment, the concentrations of Kaempferol-3-O-glucoside, Quercetin-3-O-neohesperidoside, Apigenin-6,8-di-C-glucoside-4'-O-glucoside, Quercetin-3-O-sophoroside-7-O-rhamnoside, Procyanidin C1, and Epigallocatechin were significantly greater in CK than in TH (Student’s t test, P <0.05), and the concentrations of the remaining 29 flavonoids were significantly greater in TH than in CK. 43 out of 167 phenolic acids were identified as differentially accumulated by CDs-treatment, of these 13 phenolic acids were downregulated and 30 were upregulated in TH compared with CK, among which Quillaic acid, Furanofructosyl-α-D-(6-mustard acyl)glucoside, Feruloylcaffeoyltartaric acid were found only in TH, CDs-treatment also led to a 10-fold enhancement in Syringin, Neochlorogenic acid, Homogentisic acid, and 5-fold increase in 1-Caffeoylquinic acid and 2-Caffeoylquinic acid.
Transcriptome analysis and DEGs Identification
Differentially expressed genes (DEGs) in CK vs TH comparative group were identified by a transcriptomic comparison. A total of 40.08–52.20 million clean reads were obtained, and the Q30 of the raw reads ranged from 91.08–92.21%, indicating the high quality of the transcriptome data. As shown in Figure 3A, the red dots indicate the upregulated expressed genes (Log2FC ≥ 2), the black dots indicate the non-significantly differentially expressed genes (0.5 < Log2FC < 2), and the blue dots indicate the downregulated expressed genes (fold change, Log2FC≤0.5). The horizontal axis indicates the fold change of differential expression, and the vertical axis indicates the significance level of the gene expression differences. According to the results of transcriptome data, 43340 unigenes were identified. A total of 6,874 DEGs were identified between CK vs TH (Additional files Table S3), including 2962 upregulated genes and 3912 downregulated genes (Fig. 5A).
The DEGs between CK and TH were subjected to KEGG, KOG, and GO functional pathway analyses. The top enriched KEGG terms contributed by these DEGs were ko01110 (Biosynthesis of secondary metabolites), ko04016 (MAPK signaling pathway - plant), ko00941 (Flavonoid biosynthesis), ko00860(Porphyrin and chlorophyll metabolism), and ko00909 (Sesquiterpenoid and triterpenoid biosynthesis) (Fig. 5B). The top enriched KOG terms included Posttranslational modification, protein turnover, chaperones(O), Carbohydrate transport and metabolism(G), Energy production and conversion(C), and Secondary metabolites biosynthesis, transport and catabolism(Q) (Fig. 5C). DEGs annotated in GO were classified into 52 functional groups, including 25 groups in biological process, 16 in cellular components, and 11 in molecular function. ‘Cell’, ‘Cell part’, and ‘organelle’ were the terms that dominated in the cellular component category. In the ‘molecular function’ category, the GO terms ‘catalytic activity’ and ‘binding’ predominated. ‘Cellular process’, ‘metabolic process’, ‘biological regulation’, and ‘regulation of biological process’ were the most represented GO terms in the biological process category (Fig. 5D). The above functional pathway analyses indicated that most of the identified DEGs acted on metabolic processes related to secondary metabolite metabolism and carbohydrate metabolism.
Co-expression network analysis of DEGs
To identify the candidate genes regulating secondary metabolite metabolism and carbohydrate metabolism, an effective system biology method called weighted gene co-expression network analysis (WGCNA), was performed to find the modules of highly correlated genes and relate these modules to traits. Modules are clusters of genes with high correlation, and genes of a same module are co-expressed. A total of 8 gene modules were established on the clustering and signature analysis of the genes with similar expression patterns in DEGs (Additional files: Fig. S5). They were then used to correlate with the traits of flavonoid metabolites content (Additional files: Fig. S6), phenolic acids metabolites content (Additional files: Fig. S7), and photosynthetic efficiency (Additional files: Fig. S8), respectively. Among the 8 modules, the modules of “darkturquoise” and “green” were found to be associated with flavonoid, and phenolic acids accumulation. The “darkturquoise” gene module showed a strong correlation with the concentration of flavonoids, such as isorhamnetin-3-O-gallate (neohesperidin) (r = 1.0, P < 0.01), Quercetin-4′-O-glucuronide (r = 0.98, P < 0.01), Dihydrokaempferol-3-O-glucoside (r = 0.98, P < 0.01), Quercetin-3-O-(2''-O-galactosyl) glucoside (r = 0.98, P < 0.01), Dihydrokaempferol-3-O-glucoside (r = 0.98, P < 0.01), Luteolin-7-O-glucoside (Cynaroside) (r = 0.98, P < 0.01), Kaempferol-3-O-sulfonate (r = 0.92, P < 0.01), Apigenin-7-O-rutinoside (Isorhoifolin) (r = 0.97, P < 0.01), and so on. The “green” gene module also showed a high positive correlation with flavonoids, such as Kaempferol-3-O-glucoside (Astragalin) (r = 0.95, P < 0.01), Luteolin-8-C-glucoside (Orientin) (r = 0.98, P < 0.01), Apigenin-8-C-Arabinoside (r = 0.94, P < 0.05), and so on. The “darkturquoise” gene module showed a strong correlation with the concentration of phenolic acids, such as Syringin (r = 1.0, P < 0.01), 3,4-Dihydroxybenzeneacetic acid (r = 0.99, P < 0.01), Sinapaldehyde-4-O-Glucoside (r = 0.99, P < 0.01), 2-O-Caffeoylmalic acid (r= 0.93, P < 0.01), Neochlorogenic acid (r= 0.97, P < 0.01), Homogentisic acid (r = 0.94, P < 0.01), and so on.
The module–trait relationship analysis also identified module “darkturquoise” as most highly related to photosynthetic efficiency. Photosynthetic rate (Pn) (r = 0.97, P < 0.01), stomatal conductance (Cond) (r = 0.96, P < 0.01), transpiration rate (Tr) (r = 0.99, P < 0.01), chlorophyll fluorescence parameters (Fv/Fm) (r = 0.98, P < 0.01). Therefore, the “darkturquoise” and “green” modules were selected as the key gene modules for subsequent analysis.
Key gene module expression and functional analysis
Gene annotation and expression of all the gene members in the two key gene modules were performed. “darkturquoise” and “green”gene modules were composed of 6596 and 940 unigenes, respectively. When the differential genes of CK vs TH were separately intersected with the gene members of the two key gene modules, we found that the intersection was observed between the two key gene modules and the differentially expressed genes (Fig. 6A). Downregulated expressed genes in “darkturquoise” and “green” gene modules were 985 and 83, while upregulated expressed genes in the two modules were 1621 and 44, respectively. Further expression analysis of gene members of the two key gene modules revealed distinct expression patterns in CK and TH (Fig. 6B and 6C).
The KEGG gene-set enrichment analysis of these DEGs in “green” and “darkturquoise” gene modules were severally shown as Fig. 7 and Fig. 8, respectively. For each screened gene module, the correlation scatter plot between some phenotypes and modules were drawn (Fig. 7A and Fig. 8A). The enriched KEGG terms of upregulated expressed genes in “green” gene module contained MAPK signaling pathway, porphyrin and chlorophyll metabolism, and galactose metabolism (Fig. 7B). The enriched KEGG terms of downregulated expressed genes in “green” gene module contained flavonoid biosynthesis, other glycan degradation, glycosaminoglycan degradation (Fig. 7C). The top enriched KEGG terms of upregulated expressed genes in “darkturquoise” gene module were benzoxazinoid biosynthesis, MAPK signaling pathway, porphyrin and chlorophyll metabolism, and anthocyanin biosynthesis (Fig. 8B). The top enriched KEGG terms of downregulated expressed genes in “darkturquoise” gene module were flavonoid biosynthesis, biosynthesis of secondary metabolites (Fig. 8C).
Identification of hub genes within network modules
Two modules, “darkturquoise” and “green”, were considered to take a decisive factor in the regulation of CDs on plant growth and secondary metabolites accumulation. Hub genes were defined as these genes which were highly associated with other genes in each module network, and played a central role within the network clusters. Hub genes within the two modules were discovered in Fig. 9. Protein-protein interaction (PPI) network, which took the gene members in the two key modules as its object, were obtained using string database, and a total of 44 nodes, 44 edges were identified (Fig. 9A). The MCC algorithm of CytoHubba was used to score and sort the hub nodes in the PPI network, and select the top 5 genes (triosephosphate isomerase, mitochondrial carrier protein, thymidylate kinase, dehydrogenase E1 component and lyase) as the hub genes of the PPI network (Fig. 9B). Gene expression of all PPI network gene members in the two key gene modules were performed and shown in Fig. 9C. Among them, 17 up-regulated genes and 27 down-regulated genes were identified. These interactions among key genes, pathways, metabolic type and gene regulation are shown in Fig. 9D. As for secondary metabolism, transferase family genes in anthocyanin, phenylpropanoid, and flavonoid biosynthesis pathways tended to be down-regulated after CDs-treatment, and the expression of NAD dependent epimerase/dehydratase family genes in phenylpropanoid, flavonoid, and isoflavonoid biosynthesis pathways were restrained as well. As for primary metabolism, some gene families of carbohydrate metabolism, such as pfkB family carbohydrate kinase, glycosyl hydrolase family, triosephosphate isomerase, and carbohydrate phosphorylase, tended to be down-regulated after CDs-treatment. However, some gene families involved in carbohydrate synthesis, such as sucrose synthase and aldehyde dehydrogenase families, were up-regulated. Ferrochelatase and chlorophyll A-B binding protein, which were considered to be vital components of photosynthesis and chloroplast synthesis, were both up-regulated in response to CDs-treatment.
Validation of hub genes expression by qRT-PCR
We selected the top 5 hub genes (triosephosphate isomerase, mitochondrial carrier protein, thymidylate kinase, dehydrogenase E1 component and lyase) of the PPI network to conduct a validation experiment by qRT-PCR analysis. The patterns of RNA-Seq expressions on all the 5 hub genes were highly consistent with the qRT-PCR data, and a correlation coefficient (R2) of 95.33% was obtained (Fig. 10). Both qRT-PCR and RNA-seq analyses showed that three hub genes (triosephosphate isomerase, mitochondrial carrier protein, and dehydrogenase E1 component) showed lower level of expression after CDs-treatment. However, other two genes, thymidylate kinase and lyase showed a higher level of expression in the CDs-treatment group.
{kind=link}
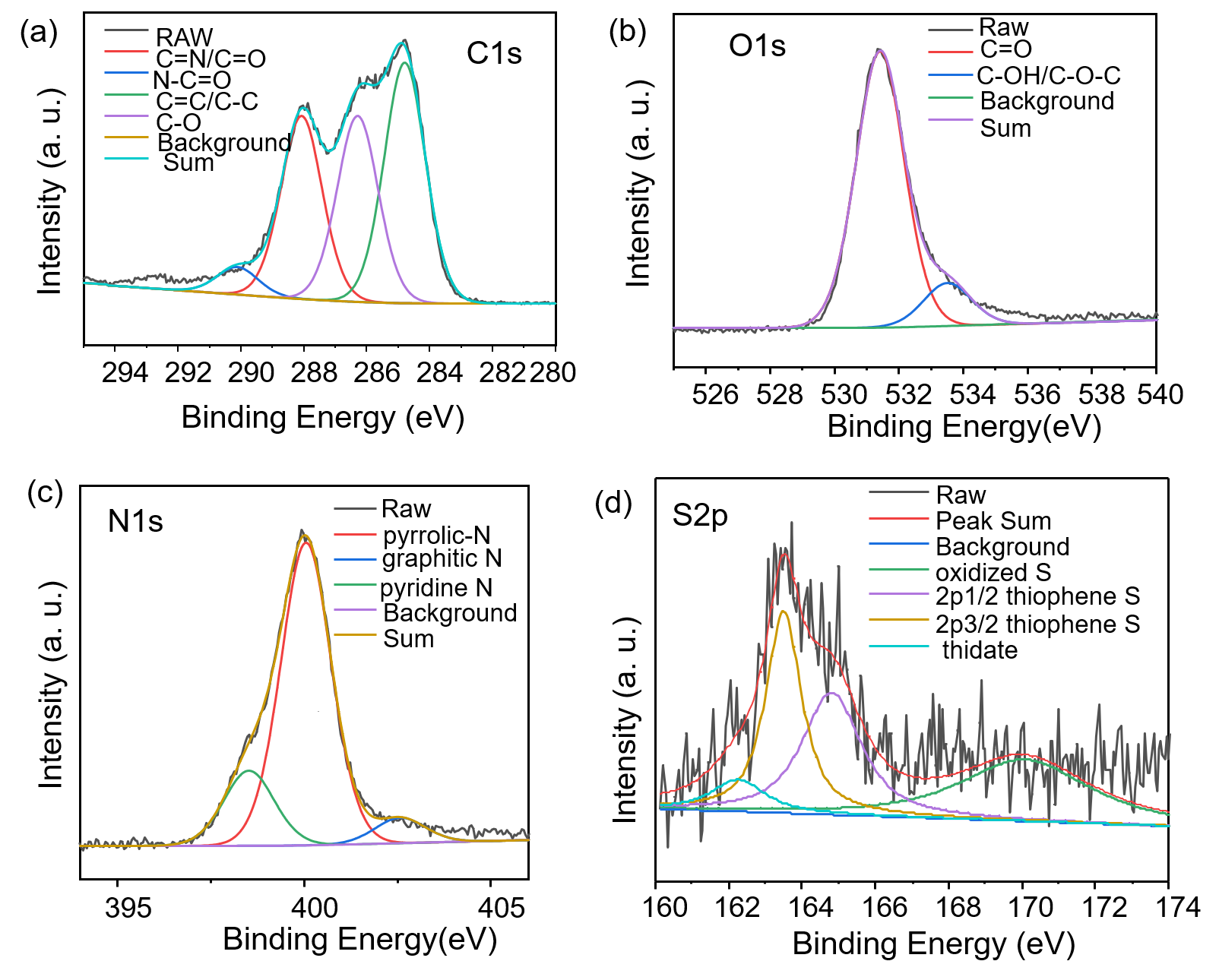{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}