Quasi-solid-state Zn-air batteries (ZABs) have shown extraordinary promise for electrochemical energy storage, but are usually limited to relatively low-rate ability (< 10 mA cm−2), which caused by the sluggish O2 electrocatalysis and unstable electrochemical interface. Here we present an ultrahigh-rate and robust quasi-solid-state ZABs integrated with the atomic Co-N4 sites anchored on wrinkled nitrogen-doped graphene (Co SA-NDGs) cathode and the modulated H-bond network of polyacrylamide (PAM) organohydrogel electrolyte with. The quasi-solid-state ZABs exhibit the highest cycling rate of 100 mA cm−2 over 50 h at room temperature, which is nearly an order of magnitude higher than results reported previously. Meanwhile, the exceptional cycling stability more than 300 h (0.5 mA cm−2) with high-capacity retention at -60 oC and all-temperature adaptability (-60 to 60°C) are also demonstrated. The integral design towards high performance Zn-air batteries using atomic site catalyst and electrolyte with good interface stability broaden the scope of application.
Article
Ultrahigh-rate quasi-solid-state Zn-air batteries enabled by atomically dispersed Co site electrocatalyst and organhydrogel electrolyte
https://doi.org/10.21203/rs.3.rs-1096489/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 27 Jun, 2022
Version 1
posted
You are reading this latest preprint version
Zn-air batteries
Co SA-NDGs
polyacrylamide organohydrogel electrolyte
- O2 reduction/evolution activity of Co-N4 sites supported on the interconnected porous nitrogen-doped graphene (Co SA-NDGs) is enhanced by curvature, contributing to decrease discharging/charging polarization and
- The introduction of dimethyl sulfoxide (DMSO) into polyacrylamide (PAM) hydrogel electrolyte effectively modulates the H-bond network, enabling improved interface stability and all-temperature adaptability (from -60 to 60oC) simultaneously.
- The quasi-solid-state Zn-air batteries with Co SA-NDGs not only achieve the record-high cycling rates of 100 mA cm-2 over 50 h at room temperature, but also show exceptional cycling stability with capacity retention over 90% more than 300 h (0.5 mA cm-2) at -60 oC.
Zn-air batteries (ZABs) with merits of high theoretical energy density (1086 Wh kg−1), environmental friendliness and abundant anode material (3.00 $ per kgZn), have attracted tremendous attention 1–3 . Nevertheless, the critical issues for high-rate and durable ZABs are as follow: (i) bifunctional O2 electrocatalyst for air cathode and (ii) electrochemical interface stability between Zn/gel electrolyte/air cathode. The discharging/charging process of the air cathode is highly dependent on the O2 reduction/evolution reaction (ORR/OER), respectively 4,5 . Unfortunately, the kinetics of the redox reaction of O2 are sluggish, causing substantial overpotentials. To date, although the expensive and scarce precious metals (Pt for ORR; Ru/Ir for OER) have been used to accelerate the O=O bond cleavage and formation process, the durable and highly efficient electrocatalysts are still urgently demanded 6,7 .
Single-atom catalysts (SACs) supported on the nitrogen-doped carbon possess the high electrical conductivity and the well-defined atomic structure with tunable coordination environment, showing nearly 100% catalytic efficiency8–11. Among transitional-metal (e.g., Fe, Co, Ni and Mn), the Co site electrocatalysts are the most promising alternatives for improving reversible O2 redox kinetics. Meanwhile, a high metal atom site accessibility and a fast charge (electron and ion) transport kinetic for high-rate ZABs is necessary, which could be realized by designing hierarchical porous nanoarchitecture. As known, the local coordination environment of central metal atoms significantly affects their electronic and geometric structures, determining the adsorption strength of oxygenated intermediates to atomic metal sites12–17. Compared to flat geometry, the curved nanostructures of electrocatalysts can lead to a large electric field enhancement to accelerate the reaction kinetics18,19.
On the other hand, the electrochemical interface stability (Zn dendrites, side reaction and corrosion, etc) greatly affects the cycling life for high-rate quasi-solid-state ZABs, which deserves enormous attention20,21. Currently, the conventional alkaline hydrogel electrolyte causes severe passivation of the Zn anode, leading to poor Zn irreversibility and limited cycling rate capability. In response, adjusting the Zn2+ solvation sheath structure and suppressing the water molecule performance could resolve the irreversibility issue. Furthermore, in extreme low-temperature, the charge transfer rate sharply decreases, which leading to the severe polarization and even invaliding batteries22–24. The anti-freezing and compatible gel electrolyte enable the adaptability at extreme condition25,26. However, limited efforts have been paid to exploit the hydrogel electrolytes for high-rate and temperature-adaptive ZABs. As an organic solvent with strong polarity, dimethyl sulfoxide (DMSO) can be used as additive to modulate the H-bond network of electrolyte. The organohydrogel electrolyte synthesized in DMSO/H2O binary solvent systems may provide a great opportunity to achieve a quasi-solid-state ZABs’ performance breakthrough.
Here we demonstrate a unprecedent quasi-solid-state ZABs that exhibit ultrahigh cycling rate of 100 mA cm−2, a long cycle life (100 h at 50 mA cm−2 and 50 h at 100 mA cm−2), and a wide operating temperature range from -60°C to 60°C. The highly wrinkled graphene creates a large charge gradient around Co-N4 site, contributing to lower the adsorption of oxygenated intermediates. Moreover, tailoring H-bond network of organohydrogel electrolyte address the interfacial stability and low-temperature-tolerance limitation. Our work simultaneously achieves an ultrahigh-rate ability at room temperature and extreme-low-temperature performance, which is a significant advance for promoting quasi-solid-state ZABs’ development toward practical applications as needed.
Synthesis and characterization of Co SA-NDGs. The synthetic procedure of Co SA-NDGs was described in Supplementary Fig. 1, including electrostatic adsorption, hydrothermal treatment, subsequent pyrolysis and acid leaching. Scanning electron microscopy (SEM, Supplementary Fig. 2) image shows that Co SA-NDGs possesses a 3D interconnected porous architecture with a highly wrinkled surface, delivering a specific surface area of 396.2 m2 g−1. The small mesopore centered at about 2~4 nm is conductive to balance the electrolyte permeation and the ion diffusion27. X-ray diffraction (XRD) pattern and Raman spectra suggest a lower graphitization degree of Co SA-NDGs (Supplementary Fig. 3 and 4, respectively)9,16. Meanwhile, a strong electron paramagnetic resonance (EPR) signal affirms the presence of plenty of defects (e.g., dangling bonds and vacant atom sites) (Supplementary Fig. 5). Transmission electron microscopy (TEM) image in Fig. 1a shows that the graphene nanosheet is highly curved. The high-curvature carbon lattices and the wrinkled nanosheets are clearly observed for Co SA-NDGs in high-resolution TEM images (Fig. 1b and Supplementary Fig. 6a-c). The ring-like selected area electron diffraction (SAED) pattern signifies the poor crystallinity. The nanosheet structure is further confirmed by atomic force microscopy (AFM) analysis with an average thickness of 3.24 nm (Supplementary Fig. 7). Aberration-corrected high angle annular dark-field scanning transmission electron microscope (AC HAADF-STEM) image distinctly detects the monodispersed bright dots (marked by yellow cycles) at atomic level (Fig. 1c), corresponding to the atomically dispersed Co atoms. The elemental mapping images of Co SA-NDGs (Supplementary Fig. 6d) verify the uniform distribution of Co, N, O and C species.
The electronic structure and local atomic configuration of Co SA-NDGs were studied by X-ray photoelectron spectroscopy (XPS) in Supplementary Fig. 8. The N 1s spectrum (Fig. 1d) is divided into pyridinic-N (398.2 eV), Co-Nx (399.1 eV), pyrrolic-N (399.9 eV), graphitic-N (400.1 eV), oxidized-N (402.1 eV), respectively. The abundant defects and nanopore make more N atoms occupy the edges, resulting in the formation of high percentage of pyridinic-N28. For the N-doped graphenes (NDGs), the positive shift (ཞ0.2 eV) of C-N bond in C 1s spectrum (Fig. 1e) of Co SA-NDGs is associated with a strong charge transfer effect between single Co atoms and curved graphene supports. The decreased electron density of C atoms and the atomic Co-Nx-C interface effectively facilitate the adsorption of intermediates and tailor the reaction energy barrier29. The loading content of Co is determined as 0.14 wt.% by inductively coupled plasma optical emission spectrometry (ICP-OES). Furthermore, the Co K-edge of Co SA-NDGs in X-ray absorption near-edge structure (XANES) exhibits an increased white-line intensity compared to that of Co foil (Supplementary Fig. 9), suggesting the valence state of Co atoms is positive. The extended X-ray absorption fine structure (EXAFS) spectra (Fig. 1f) display two peaks located at ~1.4 Å and 1.9 Å respectively, which can be assigned to Co-N and Co-C scattering paths30,31. The coordination number of Co-N is ཞ3.8 (Supplementary Table 1), suggesting the dominant Co-N coordination in Co SA-NDGs is likely to be Co-N4 structure. Besides, no peak of the metallic Co-Co bond or other high-shell peaks for Co SA-NDGs is detected compared with Co foil, confirming the Co atoms are atomically dispersed.
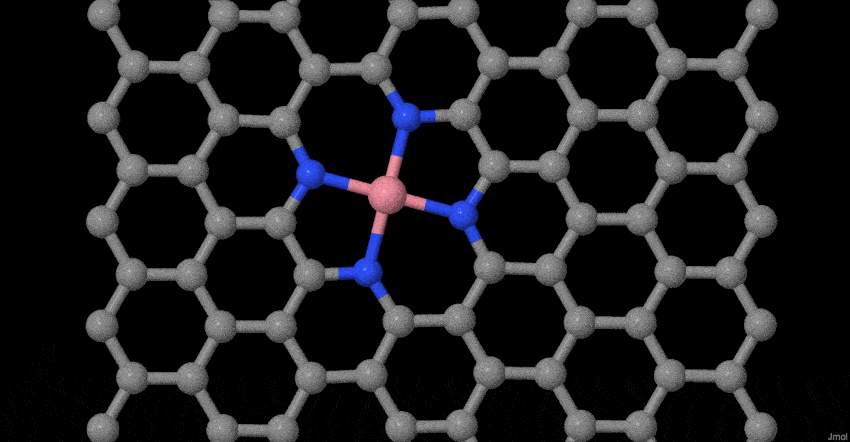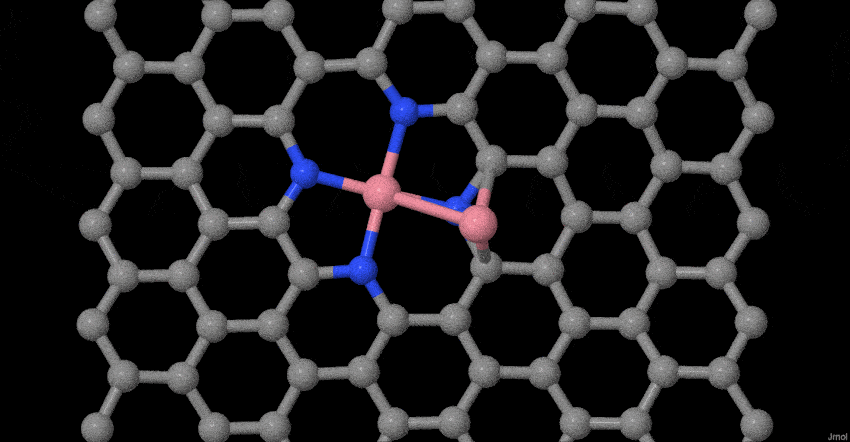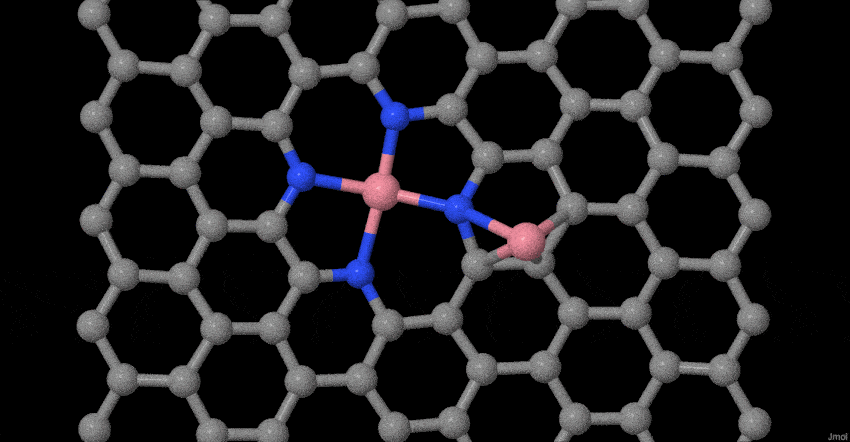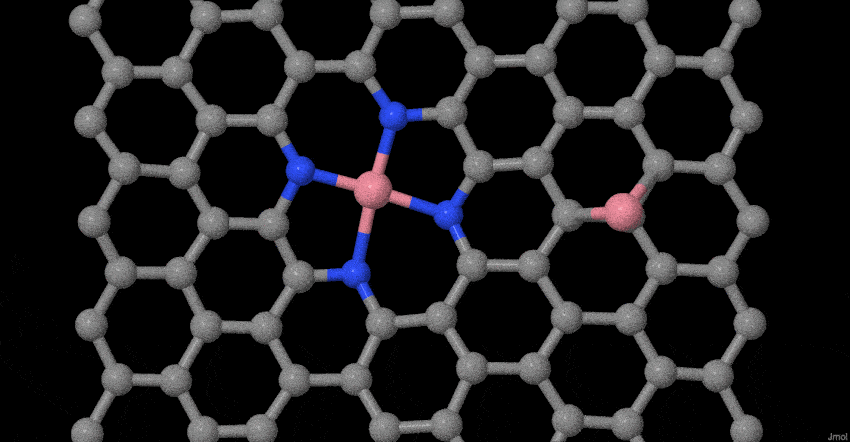
To deeply understand the evolution process of atomically dispersed Co atoms during pyrolysis, in-situ temperature-dependent TEM investigations and molecular dynamics (MD) simulations were performed. The structural evolution was timely monitored in the temperature window from 25 to 600 oC (Fig. 1g). In our case, the reduced graphene oxide (rGO) becomes less crystallization as the temperature rises to 600°C, which may be ascribed to the heteroatom doping effect32,33. More interestingly, the related Co-based nanoparticles or clusters do not appear. Furthermore, based on our previous model34, we place Co dimers on tetrapyridine N-doped carbon sites that act synergistically with vacancy defects to examine possible changes at 800 oC. The results show that two Co single atoms do not form a stable Co dimer (Fig. 1h). The Co-Co bond of the dimer gradually becomes longer until it is broken. The Co connected to the tetrapyridine N automatically forms a typical planar Co-N4 configuration, while other Co atom will become more and more away from the Co-N4 coordination. This result fully proves that stable Co dimers are not easy to form, but due to the synergistic effect of charge transfer and vacancy defects, the typical planar four-coordinate configuration of Co-N4 is formed. Simultaneously, the escaped Co atom may be captured by the doped N species or graphene defects until it is stabilized. The entire evolution process with more sets of images is supplemented (Supplementary Movie S1). These experimental observations and MD simulations provide solid evidences of the evolution process for the atomically dispersed Co-N4 structure during synthesis.
Electrocatalytic O 2 performance and DFT calculation. The O2 electrocatalytic activities of Co SA-NDGs, NDGs and commercial Pt/C are comprehensively evaluated in 0.1M KOH solution. Linear sweep voltammetry (LSV) curves (Fig. 2a) exhibit that Co SA-NDGs has an excellent ORR activity with the onset potential (Eonset) of 1.02 (V vs. RHE), superior to that of Pt/C (0.96 V). At the potential of 0.85 V, Co SA-NDGs reaches a kinetic current density (Jk) of 11.7 mA cm−2 (Fig. 2b), ~ 2.18 times higher than that of Pt/C (5.36 mA cm−2). The rapid ORR kinetic of Co SA-NDGs is supported by a small Tafel slope of 54 mV dec−1 (Supplementary Fig. 10b). The fitted Koutecky-Levich (K-L) plots calculated from the LSV curves show a good linearity (Supplementary Fig. 11), signifying that the Co SA-NDGs mainly follow 4e− ORR pathway35,36. The accelerated degradation test of Co SA-NDGs was performed by cycling catalyst between 0.6 and 1.0 V over 5000 continuous cycles (Supplementary Fig. 12a), showing little negative shift in E1/2 (∼11 mV). After injecting methanol into the electrolyte, almost no disturbance in current response is found for Co SA-NDGs, whereas a sharp decline is observed for Pt/C (Supplementary Fig. 12b). The Co species are still well distributed after durability test, evidenced by TEM and mapping results (Supplementary Fig. 13). These results confirm the remarkable ORR stability and methanol tolerance of Co SA-NDGs.
Then, the electrocatalytic OER behavior of Co SA-NDGs was also assessed. An overpotential of 350 mV for Co SA-NDGs is required to reach current density of 10 mA cm−2 (Fig. 2c), which is comparable to that of Ir/C (345 mV) and RuO2 (338 mV) but lower than that of NDGs (430 mV). Moreover, the rapid OER kinetics are elucidated by Tafel slope and electrochemical impedance spectroscopy (EIS) (Supplementary Fig. 14). Specifically, the activation energy (Ea), which is obtained from the slope of the plotted log (j) vs. 1/T, is a vital criterion for scaling the difficulty of catalytic reaction37. The experimentally measured activation energy (Ea) of Co SA-NDGs and NDGs are 28.6 and 44.0 KJ mol−1 (Fig. 2d and Supplementary Fig. 15), respectively, suggesting the favorable thermodynamic OER behavior of Co SA-NDGs. The electrochemical active surface area (ECSA) can be estimated from double layer capacitance (Cdl) because of a linear relationship34,36. The measured Cdl vale are 26.3 and 20.6 mF cm−2 for Co SA-NDGs and NDGs (Supplementary Fig. 16), corresponding to the ECSA of 657.5 and 515.0 cm−2, respectively. The high ECSA of the Co SA-NDGs represents the sufficient exposure of active sites. Generally, the bifunctional activity is evaluated by ΔE (ΔE=EOER:j=10-EORR:1/2). We use E1/2 and E10 index as the horizontal and vertical axis for better comparison, where the point located at the upper right demonstrates a better bifunctional capability38,39. The ΔE of Co SA-NDGs is only 0.71 V, ranking the top level among non-precious metal SACs and metal nanoparticle-based bifunctional catalysts (Fig. 2e and Supplementary Table S2).
The crucial role of curvature on atomic Co-N4-C system were studied using first-principles calculations based on DFT40 (Fig. 2f). Fig. 2g gives the calculated differential charge densities for the planar Co-N4 and curved Co-N4 model to show the charge transfer difference. The curved Co-N4 shows more localized charge densities and a larger charge gradient than the planar Co-N4, which is believed to facilitate the subsequent O2 activation41. Density of states (DOS) analysis (Fig. 2h) show that the d-band center of curved Co-N4 is downshifted compared to that of planar Co-N4, indicating that the adsorption of oxygenated intermediates is lowered16. Especially, the ORR activity of bifunctional electrocatalyst determines the energy conversion efficiency to a great extent. The free energy pathways of two models are both downhill on the four-electron ORR processes at U = 0 V, revealing that each elementary step can be carried out spontaneously (Supplementary Fig. 17). At U = 1.23 V, the rate-determining step is the first protonation of the adsorbed OOH* species. The ORR overpotential of curved Co-N4 site under alkaline condition is calculated to be 0.758 eV, which is lower than that of planar Co-N4 site (0.90 eV), suggesting the beneficial effect of graphene curvature for optimizing the ORR activity. Meanwhile, the stable adsorption configuration of intermediates is provided (Supplementary Fig. 18).
Aqueous ZABs with Co SA-NDGs
Then the aqueous ZABs with Co SA-NDGs and Pt/C as air cathode were assembled (Supplementary Fig. 19), in which the ambient air acted as the cathode active agent42. The open circuit voltage (OCV) and maximum power density of Co SA-NDGs are 1.53 V and 251.4 mW cm−2 (Supplementary Fig. 20a and b), respectively, which are significantly higher than these of benchmark Pt/C-based aqueous ZABs (1.48 V and 177.0 mW cm−2). The hierarchically macro/mesoporous structure is believed to facilitate rapid ion transfer and electron transport at high voltage region, thus boosting discharge ability (Supplementary Fig. 21). The Co SA-NDGs cathode delivers a high specific capacity of 757.4 mAh g−1 and an energy density of 956 Wh kg−1 at 10 mA cm−2 in Supplementary Fig. 20c, outperforming that of Pt/C-based counterpart (630.5 mAh g−1, 788 Wh kg−1). The superior rate capability is further assessed by comparing the discharge voltage platform at series of current densities (Supplementary Fig. 20d). The Co SA-NDGs cathode enables high voltage platform especially at high discharge depth over 50 mA cm−2. Notably, the ZABs with Co SA-NDGs could discharge more than 240 h at 100 mA cm−2 (Supplementary Fig. 20e). The instable interface induced by the generated Zn dendrites may account for the voltage loss at initial stage (Supplementary Fig. 20f). Unlike the Pt/C+RuO2-based ZABs with significant voltage decay, a robust discharging/charging stability (300 h, every cycle of 22 min) for Co SA-NDGs-based ZABs is achieved (Supplementary Fig. 22). Conclusively, the superior performance (e.g., maximum power density, specific capacity and cycling time) which is among the top level reported so far (Supplementary Table 3), mainly originate from atomically dispersed Co-N4 sites with curved nanostructure and the hierarchically interconnected structure of graphene architecture.
Quasi-solid-state ZABs with Co SA-NDGs
Here, we firstly synthesize and examine the physicochemical properties of PAM hydrogel electrolyte (Supplementary Fig. 23). The quasi-solid-state ZABs with Co SA-NDGs using PAM hydrogel electrolyte show a maximum power density of 219.9 mW cm−2 and a stable discharge voltage platform when varying discharging current density (2, 5 and 10 mA cm−2). The discharging voltage in rate performance is slightly higher than that of cycling measurement, which may be attributed to the interface difference43,44. However, the high-rate performance is still challenging because of the formation of dense Zn dendrites and the destruction of Zn/hydrogel electrolyte interface (Supplementary Fig. 24 and 25). Therefore, modulating the intrinsic properties of hydrogel electrolyte is particularly vital to alleviate these issues for achieving high-rate capability at ambient condition.
As known, DMSO is a favorable H-bond acceptor to form strong H-bond network with water molecules (Fig. 3a), reconstructing the solvation Zn2+ solvation sheath structure. On the one hand, the side reaction activity in aqueous electrolytes is obviously suppressed (Supplementary Fig. 26)26, 45–47. In situ optical visualization observation reveals a smooth interface of Zn/electrolyte (Fig. 3b and c) when adding DMSO into electrolyte, whereas the severe Zn dendrites appeared in DMSO-free electrolyte48. The PAM organhydrogel electrolyte synthesized in DMSO/H2O binary solvent systems retains a good stretchability (Supplementary Fig. 27). SEM images (Fig. 3d) reveal an interconnected porous structure, facilitating the electrolyte trapping and fast migration of Zn2+ ion diffusion during electrochemical reactions. In Fig. 3e, the ionic conductivities of the PAM organhydrogel electrolyte shows a high conductivity at room temperature (0.26 S cm−1) and even at -40, -60 and 60°C (0.04, 0.0087 and 47.56 S m−1, respectively), illustrating an efficient broad temperature adaptability. In order to investigate the compatibility between Zn anode and organhydrogel electrolyte, Zn/Zn symmetric batteries were constructed. The Zn/Zn symmetric batteries with PAM organhydrogel electrolyte displays a more durable stripping/plating cycles over 200 h in contrast to the PAM hydrogel electrolyte (Fig. 3f), suggesting a stable electrochemical interface at room temperature. Meanwhile, ex XRD patterns (Supplementary Fig. 28) display the relatively weak intensity of formed Zn dendrites (ZnO) when using PAM organhydrogel electrolyte. These results indicate that the introduction of DMSO effectively alleviates Zn dendrites and improves Zn/gel electrolyte interface stability via modulating the H-bond network of organhydrogel electrolyte. Notably, the quasi-solid-state ZABs with Co SA-NDGs using PAM organhydrogel electrolyte present a robust charging/discharging cycle at 50 and 100 mA cm−2 (Fig. 3g). A statistical big data analysis (Fig. 3h and Supplementary Table 4) highlights this groundbreaking result at high cycling rates, implying the promising application.
Furthermore, considering the practical demand of rechargeable batteries in cold regions, highland, etc., there is an urgent need to develop ultra-low-temperature (<-40 oC) quasi-solid-state ZABs. As far as we know, ZABs worked below -40°C is seldomly reported owing to the pronounced increase in interfacial and charge-transfer resistance when operating temperature drops from 25 to -60°C (Supplementary Fig. 29). The slow ion transport in ultra-low-temperature environment limits the depth of discharge and leads to low critical current density49. Then, the temperature-tolerance abilities of PAM organhydrogel electrolyte are further rationalized by the differential scanning calorimetry (DSC) and dynamic mechanical analysis (DMA)50,51. As shown in Fig. 4a, the PAM organhydrogel electrolyte remains transparent at -40 oC and further transforms into an opaque slurry gel at -60 oC. DSC curve shows that the freezing point of the PAM organhydrogel electrolyte is less than -70 oC without the emergence of an exothermic peak. Apart from the strong inter-molecular hydrogen bonds between DMSO and H2O, the binding energy (Eb) between the water and terminal group of PAM organhydrogel electrolytes also contributes to lower the solid-liquid transition point21,52−54. Although the Eb (-0.268 eV@PAM-W) of terminal acylamino group with neighboring water molecules via the dipole-dipole interaction is slightly higher than that of two water molecules (-0.246 eV@W-W), the Eb of alkalified acylamino group with water molecule (A-PAM-W) increases substantially to -0.379 eV (Fig. 4b). The stronger Eb represents the lower the freezing point and the better the ion migration rate. The symmetric Zn/Zn battery with the PAM organhydrogel electrolyte exhibits a stable Zn plating/stripping process over 500 h at -60°C (Supplementary Fig. 30). A rough surface and dendrite-free morphology of the cycled Zn plate (Fig. 4c) contributes to cycling stability55. Additionally, the strong inter-molecular H-bonds between DMSO and H2O result in a decrease in the saturated vapor pressure of H2O molecular, preventing the evaporation of H2O at elevated temperatures. The DMA result shows that the glass transition temperature (Tg) of the PAM organhydrogel is 125°C (Supplementary Fig. 31). These results are indicative of anti-freezing and thermally stable properties for the as-synthesized PAM organhydrogel electrolyte.
At -40°C (Supplementary Fig. 32), the critical current density of the assembled quasi-solid-state ZABs is 2 mA cm−2 under steady-state discharging test. The specific capacity still reaches 778.4 mAh g−1 at 2 mA cm−2 at -40°C, corresponding to an energy density of 918.5 Wh kg−1. A comparison between the energy density and operating temperature of the fabricated quasi-solid-state ZABs and other low-temperature batteries previously reported (Fig. 4d and Supplementary Table 5), suggests the intrinsic advantage of ZABs for low-temperature energy storage. With further decrement to -60°C, the discharge voltages at 0.1, 0.5 and 1.0 mA cm−2 are 1.30, 1.25 and 1.18 V (Fig. 4e), respectively. Fig. 4f exhibits the charging/discharging cycles at 0.5 and 1.0 mA cm−2. The long-cycle durability with capacity retention over 90% is achieved at -60°C. To the best of our knowledge, this is the record of lowest operation temperature of ZABs reported so far. Moreover, this quasi-solid-state ZABs also operate well at from 20 to 60°C (Supplementary Fig. 33). At elevated temperature of 60°C, the maximum power density is 285.7 mW cm−2 with the average discharge voltages of 1.26 V @ 10 mA cm−2 and 1.23 V @ 20 mA cm−2, respectively. The remarkable cycling stability is also recorded without significant decay after 60 h.
In summary, the ultrahigh-rate quasi-solid-state ZABs were constructed. On the one hand, the curvature enhances the O2 adsorption on Co-N4 sites and lowers the oxygenated intermediates. Also, the highly interconnected graphene network with hierarchical porous structure of Co SA-NDGs is essential for achieving fast charge transport and high-rate cycling. On the other hand, the PAM organhydrogel electrolyte mitigate Zn dendrite formation and suppress side reaction, significantly increasing the electrochemical interface stability via modulating cationic solvation structure. Consequently, at cycling rates as high as 100 mA cm-2, the quasi-solid-state ZABs achieves a record-level performance at room temperature, exceeding almost all known results. Benefiting from the anti-freezing and thermally stable abilities of PAM organhydrogel electrolyte, the quasi-solid-state ZABs still show the robust cycling stability with high-capacity retention at -60 and 60 oC. This work may pave the way for achieving high-rate batteries by integral design, which is expected to be general for other electrochemical devices.
Materials synthesis. For the synthesis of Co SA-NDGs, a certain amount of Co(NO3)2·6H2O was added into 30 mL of graphene oxide aqueous solution (2 mg mL-1) for ultrasonic 1 h. Then, 300 mg of g-C3N4 nanosheets were added into the above solution with vigorous stir for 2 h at room temperature. This precursor solution was transferred into autoclave (50 mL) and then kept at 180 °C for 12 h. After cooling, a black hydrogel was freeze-dried for 72 h to remove excess H2O while maintaining its pristine morphology. The dried cylindric aerogel was placed in a tube furnace and then heated to 800 °C (3 °C min-1) for 3 h under flowing N2 protection, followed by cooling to room temperature. The as-prepared sample was etched in 1 M HCl solution at 80 oC for 48 h. The sample was subsequently washed with water for five times and finally dried at 60 °C for overnight. NDGs was prepared with the same synthesis procedure of Co SA-NDGs except that without Co(NO3)2·6H2O introduction.
Synthesis of PAM hydrogel electrolyte. The PAM hydrogel electrolyte was prepared as follow: 4 g of acrylamide, 4 mg of methylenebisacrylamide and 10 mg of potassium persulfate were dissolved in 10 mL of Milli-Q water under magnetic stirring. The resultant solution was poured into a transparent box. Then the box was sealed with tape and placed in an oven at 60 oC for overnight. The as-prepared PAM hydrogel was immersed in 6 M KOH + 0.2 M Zn(Ac)2 electrolyte for 72 h before use.
Synthesis of PAM organhydrogel electrolyte. The PAM organhydrogel electrolyte was prepared as follow: 4 g of acrylamide, 4 mg of methylenebisacrylamide and 10 mg of potassium persulfate were dissolved in 10 mL mixed solvent (5 mL of Milli-Q water and 5 mL of DMSO) under magnetic stirring at room temperature. The resultant solution was poured into a transparent box. Then the box was sealed with tape and placed in an oven at 60 oC for overnight. The as-prepared PAM organhydrogel was immersed in 6 M KOH + 0.2 M Zn(Ac)2 electrolyte for 72 h before use.
Materials characterization. Field-emission SEM (Hitachi S-480, Japan) was performed to investigate the morphology. TEM, high-resolution TEM (JEOL JEM-2100F, Japan) and elemental mappings were conducted to characterize microstructure. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images were collected by using a JEOL 200F transmission electron microscope operated at 200 keV, equipped with a probe spherical aberration corrector. Atomic force microscopy (AFM) was carried out on a Nanonavi E-Sweep N environment control scanning probe microscope. Powder X-ray diffraction (XRD) patterns were recorded on a Bruker AXS D8 Advance device using Cu-Kα radiation (λ = 1.5418 Å). Raman spectra were collected on a Renishaw in Via Raman spectrometer with the aser wavelength of 532 nm. XPS experiments were conducted on a RBD upgraded PHI-5000C ESCA system (Perkin Elmer) with Mg Kα radiation (h=1253.6 eV). The Brunauer-Emmett-Teller (BET) specific surface area was measured by nitrogen adsorption at 77 K on a surface area and porosity analyzer (Micrometrics ASAP 2020). FTIR spectra were obtained on a FTIR spectrometer (Nicolet 6700, Thermo Electron Scientific Instruments) to record the vibration or stretch of the functional groups in the gel electrolyte. The stretchable properties were investigated using Q800 DMA (TA Instruments). The freezing point of PAM organhydrogel electrolyte was characterized by a DSC1 (Mettler Toledo) under the N2 atmosphere with a cooling rate of 3 °C min-1. The ion conductivity was determined by A.C. Impedance analyzer (E4990A, USA) on a four-electrode cell.
XAFS measurements and analysis details. The X-ray absorption fine structure spectra (Fe K-edge) were collected at 1W1B station in Beijing Synchrotron Radiation Facility (BSRF). The storage rings of BSRF were operated at 2.5 GeV with an average current of 250 mA. Using Si(111) double-crystal monochromator, the data collection was carried out in transmission/fluorescence mode using ionization chamber. All spectra were collected in ambient conditions. The acquired EXAFS data were processed according to the standard procedures using the ATHENA module implemented in the IFEFFIT software packages. The k3-weighted EXAFS spectra were obtained by subtracting the post-edge background from the overall absorption and then normalizing with respect to the edge-jump step. Subsequently, k3-weighted χ(k) data of Co K-edge were Fourier transformed to real (R) space using a hanning windows (dk=1.0 Å-1) to separate the EXAFS contributions from different coordination shells. To obtain the quantitative structural parameters around central atoms, least-squares curve parameter fitting was performed using the ARTEMIS module of IFEFFIT software packages.
Electrochemical measurement. The electrochemical measurements were performed in a three-electrode system (CHI 660e workstation, Chenhua, China) in 0.1 M KOH at room temperature. SCE electrode with saturated 3 M KCl solution and graphite rod acted as reference and counter electrodes, respectively. LSV curves were recorded after 90% IR-compensation. All potentials were converted to the RHE scale according to E(RHE)=E(SCE) + (0.241 + 0.059 pH) V. Rotating ring disk electrode (RRDE-3A, ALS, U.S.A) loaded with catalyst ink was used as the working electrode. To prepare the catalyst ink, 6 mg catalysts and 40 μL of 5 wt% Nafion solution (5%, D520) were dispersed in 960 μL water-isopropanol solution (volume ratio of 3:1) by sonicating for 1 h to form a homogeneous ink. Then 10 μL of catalyst ink was dropped onto the glass carbon electrode, and dried at room temperature. The samples were firstly activated in N2-saturated 0.1 M KOH by CV method (scan rate of 50 mV s-1) to reach a stable condition. Before ORR test, the O2 was bubbled into electrolyte to maintain O2 saturation during electrocatalytic ORR process. LSV curves were recorded in O2-saturated 0.1 M KOH with a sweep rate of 5 mV s-1 ranging from 400 to 1600 rpm.
The 4e- pathway of catalysts was estimated by the following equation:

Where j is the measured current density; jk and jL represent the kinetic- and diffusion-limiting current densities, respectively; ω is the disc rotation angular velocity of the disk, n represents calculated number of transferred electron in ORR; F is the Faraday constant (F =96485 C mol-1); C0 is the bulk concentration for O2 (1.2×10-6 mol cm-3) dissolved in 0.1 M KOH solution; D0 is the diffusivity of O2 (1.9×10-5 cm2 s-1); ν is the kinematic viscosity of electrolyte, and k is the electron-transferred rate constant.
For OER experiments, LSV curves were obtained at a scan rate of 5 mV s-1. The double layer capacitance (Cdl) was determined by CV measured at different scan rates in the non-faradaic region. The capacitive current measured at 1.10 V was plotted as a function of scan rate. EIS was measured in the frequency range from 105 to 0.001 Hz at 1.55 V. The accelerated degradation tests were performed by continuous CV cycles with a scan rate of 100 mV s-1 in 0.1 M KOH. The methanol crossover effect was explored by chronoamperometric responses at 0.7 V. In this process, 5 mL of methanol was injected into O2-saturated electrolyte.
Aqueous ZABs measurement. Aqueous ZABs were assembled using the flow cell configuration, not only diluting the byproducts accumulation but also prolonging the lifetime of batteries. The design of air electrode adopted a sandwich-like structure, which is composed of catalyst layer, water-proof breathable membrane and Ni foam layer. The preparation of catalyst layer was as follow: sufficiently mix the catalyst (60 mg) and acetylene black (10 mg) in ethanol solution,then add the polytetrafluoroethylene (PTFE) emulsion (60 wt%, 40 μL). After mixing for 30 min and drying at room temperature to remove excessive ethanol, the catalyst layer was cut into 1.0 cm × 1.0 cm pieces. Then, catalyst layer, water-proof breathable membrane and acid-pretreated Ni foam were compressed by roller press, obtaining the air electrode. The air electrode should be kept in vacuum before use. A polished Zn plate (purity 99.9 wt%, 0.3 mm thickness) was used as the anode. For rechargeable ZABs, 6M KOH + 0.2M Zn(Ac)2 was used as electrolyte. For comparison, the aqueous ZABs were also constructed with the commercial Pt/C or Pt/C+RuO2 catalyst as air electrode. The galvanostatic discharge curves were recorded by LSVs (scan rate of 5 mV s-1) without IR-compensation. Both the current density and power density were normalized to the effective surface area of air electrode. The specific capacity and energy density were calculated according the equation below:

The rate performance and discharge-charge cycling (10 min discharge, 1 min shelving, 10 min charge followed by 1 min shelving) were performed in the NEWARE testing system.
Quasi-solid-state ZABs measurement. The design of quasi-solid-state ZABs was typical sandwich-like structure. The as-prepared air electrode and Zn plate were placed on the two sides of PAM gel electrolyte. To maintain good interfacial contact during batteries measurement, the stack pressure was employed. The extreme low-temperature ZABs measurements were conducted in the low-temperature test chamber (Haier, DW-60W151EU1).
Molecular dynamics simulations. Our first principles calculations were based on density function theory (DFT) using the Perdew-Burke-Ernzerhof (PBE) form for the generalized gradient approximation as implemented in the Vienna ab initio simulation package code56-58. The projector augmented wave method with a plane-wave basis set was adopted and the energy cutoff was set to 500 eV. A Monkhorst-centered 7×7×1 k-mesh for structure optimization and total energy calculations. We set the vacuum space to 15 Å along the z-direction, as well as the convergence criteria for total energy and force were set to 10-6 eV and 0.01 eV/Å, respectively.
Quantum chemistry calculations. The computations were performed using the VASP code with PBE method. The van der Waals force was involved through the Grimme-D2 scheme in order to correctly address the adsorption of the molecules and radicals on the surface59. The model was built with a CoN4 doped graphene on top of a C60 sphere to construct a structure with curvature, and then relaxed for both the volume and the atomic geometries. Then a strain was applied in xy plane and relaxed the atomic positions for a model with larger curvature. A 20 Å vacuum was added in the z direction to exclude the possible interactions due to the periodicity. The cutoff energy of the plane wave basis set was taken as 450 eV, and only the gamma point was used for the calculations. The convergence thresholds of energy and force were set to 0.01 eV and 10-6 eV/Å, respectively. Moreover, the Coulomb interaction strengths (U and J parameters) were self-consistently achieved with a local screened Coulomb correction (LSCC) approach60, and the interactions were incorporated through a DFT+U scheme61.
Adsorption energy calculations. In adsorption energy calculations, Density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP), based on projector-augmented-wave (PAW) potential. The Perdew–Burke-Ernzeralized gradient approximation (GGA) using the approach of Grimme (DFT-D3) is used to describe the van der Waals force (vdW) between atoms. All structural models adopt cut-off energy of 500 eV, choose 2Monkhorst–Pack method as the Brillouin zone integral calculation scheme. The convergence criterion for self-consistent field iteration is 1×10−6 eV; the convergence criterion for structural optimization is 0.01 eV/Å. The adsorption energy 𝐸𝑎𝑑𝑠 can be calculated by the following equation:
𝐸𝑎𝑑𝑠 = 𝐸𝑡𝑜𝑡𝑎𝑙 – 𝐸polymer– 𝐸water
where 𝐸𝑡𝑜𝑡𝑎𝑙 is the total energy, 𝐸polymer is the energy of the polyme, and 𝐸water is the energy of the water. The higher absolute value of 𝐸 indicates stronger interactions.
- Wu, K., Zhang, L. & Yuan, Y. et al. An iron-decorated carbon aerogel for rechargeable flow and flexible Zn-air batteries. Adv. Mater. 32, 2002292–2002300 (2020).
- Shinde, S. S., Jung, J. Y. & Wagh, N. K. et al. Ampere-hour-scale zinc-air pouch cells. Nat. Energy 6, 592–604 (2021).
- Lei, Y., Wang, Q. & Peng, S. et al. Electrospun inorganic nanofibers for oxygen electrocatalysis: design, fabrication, and progress. Adv. Energy Mater. 10, 1902115–1902143 (2020).
- Hu, B., Huang, A. & Zhang, X. et al. Atomic Co/Ni dual sites with N/P-coordination as bifunctional oxygen electrocatalyst for rechargeable zinc-air batteries. Nano Res. 14, 3482–3488 (2021).
- Tang, T., Jiang, W. J. & Liu, X. Z. et al. Metastable rock salt oxide-mediated synthesis of high-density dual-protected M@NC for long-life rechargeable zinc-air batteries with record power density. J. Am. Chem. Soc. 142, 7116–7127 (2020).
- Sun, W., Wang, & F. Zhang, B. et al. A rechargeable zinc-air batteries based on zinc peroxide chemistry. Science 371, 46–51 (2021).
- Luo, M., Zhao, Z. & Zhang, Y. et al. PdMo bimetallene for oxygen reduction catalysis. Nature 574, 81–85 (2019).
- Wang, Y., Chu, F., Zeng, J., Wang, Q. & Naren, T. Single atom catalysts for fuel cells and rechargeable batteries: principles, advances, and opportunities. ACS Nano 15, 210–239 (2021).
- Chen, Y., Gao, R. & Ji, S. et al. Atomic-level modulation of electronic density at cobalt single-atom sites derived from metal-organic frameworks: enhanced oxygen reduction performance. Angew. Chem. Int. Ed. 60, 3212–3221 (2021).
- Ji, D. Fan, L. & Li, L. et al. Atomically transition metals on self-supported porous carbon flake arrays as binder-free air cathode for wearable zinc-air batteries. Adv. Mater. 31, 1808267–1808274 (2019).
- Li, S., Cheng, C., Zhao, X., Schmidt, J. & Thomas, A. Active salt/silica-templated 2D mesoporous FeCo-Nx-carbon as bifunctional oxygen electrodes for zinc-air batteries. Angew. Chem. Int. Ed. 57, 1856–1862 (2018).
- Li, X., Rong, H., Zhang, J., Wang, D. & Li, Y. Modulating the local coordination environment of single-atom catalysts for enhanced catalytic performance. Nano Res. 13, 1842–1855 (2020).
- Wang, Y., Liu, Y. & Liu, W. et al. Regulating the coordination structure of metal single atoms for efficient electrocatalytic CO2 reduction. Energy Environ. Sci. 13, 4609–4624 (2020).
- Han, A., Wang, X. & Tang, K. et al. An adjacent atomic platinum site enables single-atom iron with high oxygen reduction reaction performance. Angew. Chem. Int. Ed. 60, 19262–19271 (2021).
- Liu, D., Li, X. & Chen, S. et al. Atomically dispersed platinum supported on curved carbon supports for efficient electrocatalytic hydrogen evolution. Nat. Energy 4, 512–518 (2019).
- Yang, J., Li, W. & Tan, S. et al. The electronic metal-support interaction directing the design of single atomic site catalysts: achieving high efficiency towards hydrogen evolution. Angew. Chem. Int. Ed. 60, 19085–19091 (2021).
- Jing, H., Zhu, P. & Zheng, X. et al. Theory-oriented screening and discovery of advanced energy transformation materials in electrocatalysis. Adv. Powder Mater. DOI:10.1016/j.apmate.2021.10.004, (2021).
- Yang, J., Wang, Z. & Huang, C. X. et al. Compressive strain modulation of single iron sites on helical carbon support boosts electrocatalytic oxygen reduction. Angew. Chem. Int. Ed. 60, 22722–22728 (2021).
- Gao, F., Hu, S. J. & Zhang, X. L. et al. High-curvature transition metal chalcogenide nanostructures with profound proximity effect enable fast and selective CO2 electroreduction. Angew. Chem. Int. Ed. 59, 8706–8712 (2020).
- Song, Z., Ding, J. & Liu, B. et al. A rechargeable Zn-air batteries with high energy efficiency and long life enabled by a highly water-retentive gel electrolyte with reaction modifier. Adv. Mater. 32, 1908127–1908136 (2020).
- Gu, C., Xie, X. Q. & Liang, Y. et al. Small molecule-based supramolecular-polymer double-network hydrogel electrolytes for ultra-stretchable and water proof Zn-air batteries working from -50 to 100°C. Energy Environ. Sci. 14, 4451–4462, (2021).
- Pei, Z., Yuan, Z. & Wang, C. et al. A flexible rechargeable zinc-air batteries with excellent low-temperature adaptability. Angew. Chem. Int. Ed. 59, 4793–4799 (2020).
- Zhao, C. X., Liu, J. N. & Yao, N. et al. Can aqueous zinc-air batteries work at sub-zero temperatures? Angew. Chem. Int. Ed. 60, 15281–15285 (2021).
- Zhang, Q., Ma, Y. & Lu, Y. et al. Modulating electrolyte structure for ultralow temperature aqueous zinc batteries. Nat. Commun. 11, 4463–4472 (2020).
- Liu, Z., Luo, X., Qin, L., Fang, G., Liang, S. Progress and prospect of low-temperature zinc metal batteries, Adv. Powder Mater. DOI: 10.1016/j.apmate.2021.10.002, (2021).
- Lu, C. & Chen, X. All-temperature flexible supercapacitors enabled by antifreezing and thermally stable hydrogel electrolyte. Nano Lett. 20, 1907–1914 (2020).
- Zhou, T. Shan, H. & Yu, H. et al. Nanopore confinement of electrocatalysts optimizing triple transport for an ultrahigh-power-density zinc-air fuel cell with robust stability. Adv. Mater. 22, 2003251–2003258 (2020).
- Yia, S., Qin, X. & Liang, C. et al. Insights into KMnO4 etched N-rich carbon nanotubes as advanced electrocatalysts for Zn-air batteries, Appl. Catal. B: Environ. 264, 118537–118546 (2020).
- Shinde, S. S., Lee, C. H. & Jung, J. Y. et al. Unveiling dual-linkage 3D hexaiminobenzene metal-organic frameworks towards long-lasting advanced reversible Zn-air batteries. Energy Environ. Sci. 12, 727–738 (2019).
- Zhang, L., Jia, Y. & Gao, G. et al. Graphene defects trap atomic Ni species for hydrogen and oxygen evolution reactions. Chem 4, 285–297 (2018).
- Zhu, Y., Sun, W. & Luo, J. et al. A cocoon silk chemistry strategy to ultrathin N-doped carbon nanosheet with metal single-site catalysts. Nat. Commun. 9, 3861–3869 (2018).
- Zhang, E., Wang, T. & Yu, K. et al. Bismuth single atoms resulting from transformation of metal-organic frameworks and their use as electrocatalysts for CO2 reduction. J. Am. Chem. Soc. 141, 16569–16573 (2019).
- Liu, D., He, Q., Ding, S. & Song, L. Structural regulation and support coupling effect of single-atom catalysts for heterogeneous catalysis. Adv. Energy Mater. 10, 2001482–2001502 (2020).
- Wang, Q., Ji, Y. & Lei, Y. et al. Pyridinic-N-dominated doped defective graphene as superior oxygen electrocatalyst for ultrahigh-energy-density Zn-air batteries. ACS Energy Lett. 3, 1183–1191 (2018).
- Gong, X., Zhu, J. & Li, J. et al. Self-templated hierarchically porous carbon nanorods embedded with atomic Fe-N4 active sites as efficient oxygen reduction electrocatalysts in Zn-air batteries. Adv. Funct. Mater. 31, 2008085–2008094 (2020).
- Chen, Z., Wang, Q. & Zhang, X. et al. N-doped defective carbon with trace Co for efficient rechargeable liquid electrolyte-/all-solid-state Zn-air batteries. Sci. Bull. 63, 548–555 (2018).
- Duan, Y., Yu, Z. Y. & Hu, S. J. et al. Scale-up synthesis of amorphous NiFeMo oxides and their rapid surface reconstruction for superior oxygen evolution catalysis. Angew. Chem. Int. Ed. 58, 15772–15777 (2019).
- Zhao, C. X., Liu, J. N. & Wang, J. et al. A ∆E = 0.63 V Bifunctional oxygen electrocatalyst enables high-rate and long-cycling zinc-air batteries. Adv. Mater. 33, 2008606–2008613 (2021).
- Yang, G., Zhu, J. & Yuan, P. et al. Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity. Nat. Commun. 12, 1734–1743 (2021).
- Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Z. et al. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 44, 2060–2087 (2015).
- Wang, Q., Ye, K. & Xu, L. et al. Carbon nanotube-encapsulated cobalt for oxygen reduction: integration of space confinement and N-doping. Chem. Commun. 55, 14801–14804 (2019).
- Jiao, D., Ma, Z. & Li, J. et al. Test factors affecting the performance of zinc-air batteries. J. Energy Chem. 44, 1–7 (2020).
- Zhao, Z., Fan, X. & Ding, J. et al. Challenges in zinc electrodes for alkaline zinc-air batteries: obstacles to commercialization. ACS Energy Lett. 4, 2259–2270 (2019).
- Aslam, M. K., Niu, Y. & Hussain, T. et al. How to avoid dendrite formation in metal batteries: innovative strategies for dendrite suppression. Nano Energy 86, 106142–106142 (2021).
- Cao, L., Li, D. & Hu, E. et al. Solvation structure design for aqueous Zn metal batteries. J. Am. Chem. Soc., 142, 21404–21409 (2020).
- Nian, Q., Wang, J. & Liu, S. et al. Aqueous batteries operated at @50 oC, Angew. Chem. Int. Ed. 58, 16994–16999 (2019).
- Nian, Q., Zhang, X. & Feng, Y. et al. Designing electrolyte structure to suppress hydrogen evolution reaction in aqueous batteries. ACS Energy Lett. 6, 2174–2180 (2021).
- Zhou, M., Guo, S. & Li, J. et al. Surface-preferred crystal plane for a stable and reversible zinc anode. Adv. Mater. 33, 2100187–2100195 (2021).
- Chen, M., Zhang, Y., Xing, G., Chou, S. L. & Tang, Y. Electrochemical energy storage devices working in extreme conditions. Energy Environ. Sci. 14, 3323–3351 (2021).
- Sun, Y., Ma, H. & Zhang, X. et al. Salty ice electrolyte with superior ionic conductivity towards low-temperature aqueous zinc ion hybrid capacitors. Adv. Funct. Mater. 31, 2101277–2101286 (2021).
- Chen, R., Xu, X. & Peng, S. et al. A flexible and safe aqueous zinc-air battery with a wide operating temperature range from -20 to 70°C. ACS Sustainable Chem. Eng. 8, 11501–11511 (2020).
- Zhao, S., Zuo, Y. & Liu, T. et al. Multi-functional hydrogels for flexible zinc-based batteries working under extreme conditions. Adv. Energy Mater. 11, 2101749–2101777, (2021).
- Zhang, Q., Luan, J. & Fu, L. et al. The three-dimensional dendrite-free zinc anode on a copper mesh with a zinc-oriented polyacrylamide electrolyte additive. Angew. Chem. Int. Ed. 58, 15841–15847 (2019).
- Pei, Z., Ding, L. & Wang, C. et al. Make it stereoscopic: interfacial design for full-temperature adaptive flexible zinc-air batteries. Energy Environ. Sci. 14, 4926–4935 (2021).
- Wang, F., Borodin, O. & Gao, T. et al. Highly reversible zinc metal anode for aqueous batteries. Nat. Mater. 17, 543–549 (2018).
- Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 54, 11169–11186 (1996).
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond, J. Comput. Chem. 29, 2044–2078 (2008)
- Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phy. Rev. Lett. 77, 3865–3869 (1996).
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
- Wang, Y. C. & Jiang, H. Local screened coulomb correction approach to strongly correlated d-electron systems. J. Chem. Phys. 150, 154116–154122 (2019).
- Liechtenstein, A. I., Anisimov, V. I. & Zaanen, J. Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators, Phys. Rev. B 52, R5467-R5471 (1995).
Acknowledgements
This work was financially supported by the Hunan Provincial Science and Technology Plan Project (2020JJ4710). Feng is supported by the Research start-up Funds (Grant No.~2019KY23) from Southwest Jiaotong University. We sincerely thank Prof. Dingsheng Wang and Prof. Yadong Li for their valuable suggestions.
Author contributions
Q.W., Y.W., P.Y., G.F., and J. L., performed the catalyst synthesis, characterizations, and electrochemical experiments, collected and analyzed the data. L.Z., and W.L., performed the XAFS measurement. Q.F., S.T., and L.X., conducted and discussed the theoretical calculations and molecular dynamics simulations. Y. L., designed the project. All authors discussed the result, and contributed to the writing of the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
There is NO Competing Interest.
- SupplementaryMovieS1.gif
Molecular dynamics simulation
- SI1.docx
SI Ultrahigh-rate quasi-solid-state Zn-air batteries enabled by atomically dispersed Co site electrocatalyst and organhydrogel electrolyte
{kind=link}