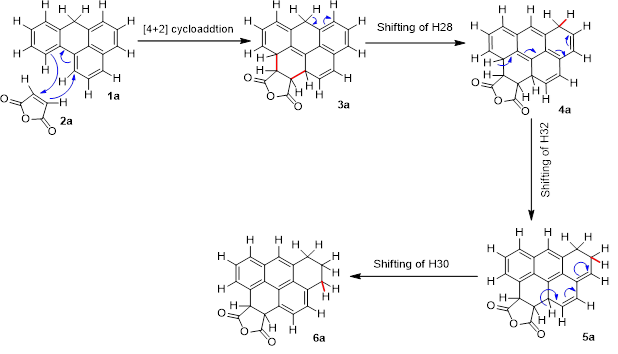
In the first step of reaction A (scheme 1), 7H-benzo[a]phenalene (1a) and maleic anhydride (2a) come close to form transition state (TS1a). Dipole moment of TS1a is 5.4 Debye which is higher than the dipole moments of the reacting species i.e., 4.4 Debye for maleic anhydride (2a) and 0.44 Debye for 7H-benzo[a]phenalene (1a). TS1a leads to the formation of an unstable intermediate 3a [40] which has a dipole moment of 5.3 Debye. In the 2nd step, H28 is transferred from C12 to C17 resulting in formation of 2nd intermediate (4a). 3rd step involves the transfer of H32 from C20 to C23 and this step needs an activation energy of 36.34 kcal/mol to form 3rd intermediate (5a). Although 5a is more stable than reactants yet it gets converted to an even more stable final product (6a) which has a dipole moment of 5.7 Debye. During transformation of 5a to 6a, H30 moves from C18 to C21. Dihedral angle of C3, C1, C2 and C5 is 0˚ in reacting species while, its value in 3a is -33.7o which further changes to -23.9o in 6a. Bond length of C1____ C2 in 2a is 1.3Å which increases to 1.5Å in 6a. Energy of all the structures relative to reactants is given in Fig. 1. In Fig. 2, structures of reactants i.e., 1a and 2a are shown along with labelling and numbering of atoms.
Table 1 elaborates the thermodynamic activation parameters and activation energy for different steps of reaction mentioned in scheme 1. The conversion of 3a into 4a via TS2a shows highest activation energy, therefore, it is the slowest step i.e. rate determining step for the reaction [54]. Free energy of activation is also maximum for this conversion. Activation energy for the first step is minimum. Thus, the formation of 3a is faster as compared to formation of any other intermediate involved in reaction. It is to be noted that activation energy and enthalpy of activation follow same trend. They decrease in following order:
step 2> step 3> step 4 > step 1
Table 1
Values of thermodynamic activation parameters for each step of scheme 1 at M06-2X/6-311++G(d,p)
|
Step Number
|
Activation energy(kcal/mol)
|
Free energy of activation(kcal/mol)
|
Enthalpy of activation(kcal/mol)
|
Entropy of activation(Cal/mol/K)
|
|
1
|
9.3
|
26.73
|
11.12
|
-52.34
|
|
2
|
75.9
|
72.14
|
71.92
|
-0.74
|
|
3
|
36.3
|
33.74
|
33.09
|
-2.16
|
|
4
|
23.0
|
22.3
|
21.79
|
-1.72
|
The trend in free energy of activation differs from these two parameters (activation energy and enthalpy). Sequence of decrease in its value is step 2> step 3> step 1> step 4
Negative values of activation entropy indicate that all the transition states have less degree of freedom as compared to the corresponding reacting species [55].
Thermodynamic properties have been used for predicting the spontaneity of chemical reactions and stability of product obtained [56]. Enthalpies, entropies and free energies (relative to reactants) of all structures involved in the scheme 1 are given in Table 2. Enthalpy of final product (6a) is -40.2 kcal/mol which indicates that the overall reaction proceeds exothermally. Apart from 6a and 5a, each structure has higher value of enthalpy compared to reactants. The first step of reaction, in which cycloaddition occurs, is endothermic and endergonic [57] step as the calculated values of ΔH and ΔG are 11.2 kcal/mol and 25.7 kcal/mol, respectively. This indicates that this step cannot occur spontaneously at the specified conditions of 298.15K and 1atm.
Values of these parameters for 2nd step are 17.7 kcal/mol and 17.46/ kcal/mol, thus this step is also endothermic and endergonic. The calculations of these values for 3rd and 4th steps revealed that these steps were exothermic and exergonic. ΔH for these steps is -39.72 kcal/mol and -29.46 kcal/mol while ΔG for them is -38.9 kcal/mol and -29.89kcal/mol respectively.
The overall entropy change in this reaction is negative, it is obvious because here two molecules are combining to form a single adduct. Decrease in entropy during formation of TS1a from two separate reactants is significantly large. Formation of other transition states also accompany negative entropy change, but the values of this change are much smaller as compared to the TS1a. This finding is consistent with the fact that TS1a is formed from two different molecules which are not covalently bonded to each other. Thus, their degree of freedom is greatly reduced which causes entropy to decrease by such a huge value [58]. Decrease in entropy is a factor that does not favors the proceeding of reaction [52].
Table 2
Values of thermodynamic parameters for scheme 1 are reported relevant to reactants at M06-2X/6-311++G(d,p)
|
Structure
|
ΔH
(kcal/mol)
|
ΔS
(Calmol−1K−1)
|
ΔG
(kcal/mol)
|
|
3a
|
11.2
|
-48.62
|
25.7
|
|
TS2a
|
83.2
|
-49.357
|
97.9
|
|
4a
|
28.9
|
-47.683
|
43.2
|
|
TS3a
|
62.1
|
-49.844
|
76.9
|
|
5a
|
-10.7
|
-50.287
|
4.3
|
|
TS4a
|
11.1
|
-52.003
|
26.6
|
|
6a
|
-40.2
|
-48.864
|
-25.6
|
All the factors mentioned in Table 1 and 2 were computed at B3LYP/6311++G(d,p) level as well. The comparison of the results of B3LYP and M06-2X functional revealed a variation among the results of these two functionals, particularly, the factors associated with 1st step (cycloaddition) showed most significant variations. According to Max Linder and Tore Brinck, M06-2X is a better choice for studying Diels Alder reaction and due to this reason, the results of only this functional have been discussed in detail [41]. Entropy is the parameter which is least affected by the change of functional.
Examining the transfer of electron density during the course of a chemical reaction, provides useful information that helps in understanding and controlling the chemical reactions [59]. To illustrate the process of electron density transfer in better way, molecular electrostatic maps (MESP) for all structures (Fig. 3) were computed. For this purpose, electrostatic potential was mapped on the self-consistent field (SCF) of total electron density [60]. MESP can be calculated directly from electron density and structural information could be obtained easily from standard quantum chemical calculations [61]. These maps display the distribution of electron density on molecules; different colors depict different values of electron density. Red color indicates the high electron density while blue color shows electron deficient site [55, 62]. MESP has been utilized for understanding many important problems in chemistry e.g. for studying site of attack by electrophile/nucleophile in a molecule and its reactivity [63]. In the 1st step of scheme 1, C20 and C18 in 1a approaches C1 and C2 of 2a. In MESP of 1a and 2a, it can be seen that C20 and C18 are electron rich sites while C1 and C2 are regions of low electron density in maleic anhydride. Thus, it is expected from their MESP that 1a is acting as nucleophile and 2a is acting as an electrophile in this reaction. This result is later confirmed by the frontier orbitals analysis of reactants which shows that reaction between 1a and 2a is normal electron demand.
Table 3
ELUMO, EHOMO and ELUMO−HOMO for all species involved in scheme 1 at M06-2X/6-311++G(d,p)
|
Stationary point
|
ELUMO (eV)
|
EHOMO (eV)
|
ELUMO−HOMO (eV)
|
|
2a
|
-2.36
|
-10.48
|
8.12
|
|
1a
|
-0.84
|
-6.85
|
6.02
|
|
TS1a
|
-1.48
|
-6.73
|
5.25
|
|
3a
|
-1.44
|
-6.83
|
5.38
|
|
TS2a
|
-1.99
|
-5.28
|
3.29
|
|
4a
|
-2.15
|
-6.03
|
3.88
|
|
TS3a
|
-1.75
|
-5.30
|
3.55
|
|
5a
|
-0.99
|
-7.22
|
6.23
|
|
TS4a
|
-1.39
|
-6.82
|
5.42
|
|
6a
|
-0.85
|
-7.36
|
6.51
|
Analysis of frontier orbitals (HOMO and LUMO) provides useful information about the reactivity of the molecules [64]. It is possible to determine electron donating ability and electron accepting abilities of molecules from EHOMO and ELUMO respectively [65]. Therefore, the energies of these orbitals are calculated for all the structures involved in the scheme 1 (Table 3). Difference in energies of LUMO and HOMO throws light on kinetic stability of the molecules. Higher value of ELUMO−HOMO indicates that structure has greater kinetic stability and it is not very reactive, whereas, lower gap in energies of frontier orbitals reveals lower stability and highly reactive nature of the molecule [64]. It is evident from Table 3 that all the transition states have relatively lower values of ELUMO−HOMO as compared to the corresponding reactants and products which is consistent with highly reactive nature of transition states [66].
By using the values of energies for frontier orbitals of diene (7H-benzo[a]phenalene) and dienophile (maleic anhydride), it can be observed whether the reaction is normal electron demand or inverse electron demand [67]. For this purpose, gap of HOMO of dienophile and LUMO of diene was calculated (Fig. 4), which is found to be 9.6eV. While gap between LUMO of dienophile and HOMO of diene is 4.5eV. As the difference of HOMOdiene and LUMOdienophile is comparatively low, thus the reaction proceeds with normal electron demand [68].
Table 4
Chemical hardness (η), softness (S), electronic chemical potential (µ), electrophilicity index (ω) and nucleophilicity index (N) for all species in scheme 1
|
Chemical Species
|
η
|
S
|
µ
|
ω
|
N
|
|
2a
|
4.06
|
0.25
|
-6.42
|
5.08
|
0.42
|
|
1a
|
3.01
|
0.33
|
-3.85
|
2.46
|
4.05
|
|
TS1a
|
2.62
|
0.38
|
-4.11
|
3.22
|
4.17
|
|
3a
|
2.69
|
0.37
|
-4.14
|
3.18
|
4.08
|
|
TS2a
|
1.64
|
0.61
|
-3.64
|
4.02
|
5.62
|
|
4a
|
1.94
|
0.52
|
-4.09
|
4.31
|
4.88
|
|
TS3a
|
1.78
|
0.56
|
-3.53
|
3.49
|
5.60
|
|
5a
|
3.11
|
0.32
|
-4.1
|
2.70
|
3.68
|
|
TS4a
|
2.71
|
0.37
|
-4.106
|
3.11
|
4.09
|
|
6a
|
3.25
|
0.31
|
-4.108
|
2.59
|
3.54
|
In Table 4, values of global reactivity descriptors are listed for all structures of scheme 1. Chemical hardness (η) basically explains structural polarization of electronic cloud and it also signifies the resistance of molecules/atoms/ions to the deformation under little perturbation during a chemical reaction. Chemical softness (S) is the reciprocal of chemical hardness and it is defined as the ability of an atom or molecule to receive the electrons. Usually, soft molecules have smaller gap between EHOMO and ELUMO as compared to hard molecules [45]. Relations used for estimation of η and S are given in computational details. Among the reactants, maleic anhydride is harder as compared to 7H-benzo[a]phenalene. Hardness of product (6a) is greater than all intermediates which shows that product (6a) is more resistant to react further. It should be noted that softness of each transition state is greater than corresponding minima structures. For example, TS4a is softer as compared to 6a and 5a.
Eq 4 has been used for the calculation of chemical potential (µ) which provided information about the charge transfer among reacting molecules [64]. As indicated in Table 4, the value of chemical potential for 7H-benzo[a]phenalene is greater than maleic anhydride which means that significant amount of charge will transferred form diene (7H-benzo[a]phenalene) to dienophile (maleic anhydride) [69, 70]. To get an idea about the abilities of molecules to accept electrons, electrophilicity index was calculated. It is one of the most significant descriptors of conceptual DFT which enables us to understand and predict physiochemical processes [71]. If value of electrophilicity index increases beyond 1.5eV for a given species then it is regarded as strong electrophile [68]. According to this principle all structures listed in Table 4 are strong electrophiles. Important point is the fact that ω is much higher for maleic anhydride (5.08eV) than 7H-benzo[a]phenalene (2.46eV) which also confirms that dienophile is acting as electrophile in this reaction [68].
Nucleophilicity index (N) which is computed by using eq 5, is related directly to polarizability and inversely to effective nuclear charge [72]. As expected, value of N is greater for diene as compared to dienophile which is also favors the interaction of HOMO of diene with LUMO of dienophil [68]. N and ω are powerful quantum chemical tools which not only tell about interaction of frontier molecular orbitals, but also predict feasibility and polar character of Diels-Alder reactions i.e. for a reaction to be more polar and faster the diene should be more nucleophilic and dinophile should be more electrophilic or vice versa [35]. Thus, from values of N and ω the polar nature of this cycloaddition reaction (between 1a and 2a) could be predicted.
Table 5
NBO charges on selected atoms of reactants and product in scheme 1 (Unit of charge is e)
| |
C1
|
C2
|
C18
|
C20
|
O4
|
|
2a
|
-0.243
|
-0.243
|
|
|
-0.564
|
|
1a
|
|
|
-0.201
|
-0.167
|
|
|
6a
|
-0.327
|
-0.337
|
-0.043
|
-0.021
|
-0.558
|
Natural bond orbital (NBO) analysis provides insight into chemical bond properties and electronic properties of molecules [73]. This analysis is directly related to electronic wave functions and is based upon unoccupied Non-Lewis and occupied Lewis localized orbitals [45]. NBO analysis provides best possible picture of natural Lewis structure, because fine details of orbitals are chosen mathematically for the inclusion of highest possible electron density [74].
As indicated in Table 5, C1 and C2 (atoms of maleic anhydride) have an equal amount of charge in the reactant, however, in the product these two atoms have slight variation in their charges. Charges on C18 and C20 changes from -0.201 and -0.167 to -0.043 and -0.021 respectively. While charges on C1 and C2 are varied from -0.243 each to -0.327 and -0.337 respectively. These values of charges on the selected atoms clearly reveal the transfer of charge from 1a to 2a [75]. Charge on oxygen atom does not show any significant variation in its value.
Fukui functions are the quantitative descriptors used to rationalize the chemical reactivity at a particular site of molecule [76]. These functions were proposed by Parr and Yang and defined as “the partial derivative of electron density with respect to total number of electrons of the system at constant external pressure” [77]. These are calculated according to eq 6 and eq 7 by using natural population.
Table 6
Condensed to atom Fukui (f+) functions for maleic anhydride (2a)
|
Atom
|
C1
|
C2
|
C3
|
C5
|
O4
|
O6
|
O7
|
|
f+
|
0.19
|
0.19
|
0.065
|
0.065
|
0.036
|
0.18
|
0.18
|
Parr and Yang also indicated that the sites in molecules which have largest values of Fukui functions are most reactive for the attack of electrophile/nucleophile [74]. The condensed to atoms Fukui functions (f+) for all carbon and oxygen atoms of 2a are calculated, results are reported in Table 6. It is clear from this table that C1 and C2 are the most reactive sites for attack of 1a (nucleophile). As 7H-benzo[a]phenalene is behaving as nucleophile, therefore, “f-” have been calculated for locating the sites of attack by electrophile. Values of these functions on all carbon atoms of 7H-benzo[a]phenalene are given in Table 7.
Table 7
Condensed to atom Fukui (f-) functions for 7H-benzo[a]phenalene (1a)
|
Atom
|
f-
|
Atom
|
f-
|
|
C10
|
0.026
|
C19
|
0.048
|
|
C11
|
0.052
|
C20
|
0.418
|
|
C12
|
0.027
|
C21
|
0.091
|
|
C13
|
0.104
|
C22
|
0.137
|
|
C14
|
0.037
|
C23
|
0.031
|
|
C15
|
0.048
|
C24
|
0.02
|
|
C16
|
0.029
|
C25
|
0.723
|
|
C17
|
0.064
|
C26
|
0.656
|
|
C18
|
0.069
|
|
|
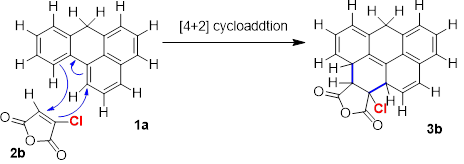
Now, the effect electron withdrawing and electron donating substituents is determined on reaction mechanism. The substituent selected as an electron withdrawing species is the chlorine. In maleic anhydride, the bond distances of C2___C5 and C1___C3 are equal i.e. 1.49Å, but their values changed to 1.487Å and 1.506Å in chloro maleic anhydride, respectively. Values of 8,1,2 and 9,2,5 bond angles also differ slightly in chloro maleic anhydride.
Introduction of chlorine on carbon 2 of maleic anhydride caused its energy to reduce by an amount of 460 Hartree. Enthalpy and free energy are also decreased by a similar amount. However, entropy in case of chloro maleic anhydride is higher than the entropy of maleic anhydride. The former has an entropy of 81 cal/mol/K, this value is 8 cal/mol/K greater than the entropy of latter. Discussion of these thermodynamic parameters indicates that the chlorine substituent has enhanced the overall stability of the structure.
Next, the effect of this substituent on the energies of frontier orbitals has been analyzed. Influence of chlorine on EHOMO and ELUMO, as one can realize by comparison of Fig. 5 and Fig. 6, is completely opposite. EHOMO is increased by 0.15eV while ELUMO is decreased by 0.19eV. ELUMO−HOMO have been reduced from 8.1eV to 7.8eV. In order to check if the reaction is still NED or it has changed to IED following energies are computed:
HOMOdienophile-LUMOdiene = 9.5eV
LUMOdienophhile-HOMOdiene=4.3eV
These values shows that not only the reaction is normal electron demand, in fact the interactions of frontier orbitals are now more favorable. Usually, it is expected that under such circumstances it becomes easier for reacting species to undergo reaction and activation barrier becomes lower. But this is not the case in study of present reaction. From Table 8, it can be seen that activation barrier became considerably high by the substitution of chlorine on maleic anhydride.
Table 8
Thermodynamic parameters for reaction B (scheme 2) at M06-2x++(d,p) level
|
Structure
|
ΔH
(kcal/mol)
|
ΔS
(Calmol−1K−1)
|
ΔG
(kcal/mol)
|
ΔE
(kcal/mol)
|
|
1a+2b
|
0
|
0
|
0
|
0
|
|
TS1b
|
38.12
|
-47.95
|
52.42
|
38.4
|
|
3b
|
9.63
|
-51.66
|
25.03
|
7.4
|
This unusual behavior can be attributed to the much higher stability of chloro maleic anhydride as compared to the maleic anhydride as discussed above. According to L.J. Andrews and R.L Keefer following resonance structure is responsible for this increased stability of chloro maleic anhydride [23]:
Another possible reason for the elevated reaction barrier of reaction B as compared to reaction A, could be the increase in steric hindrance due replacement of hydrogen by the chlorine [78].
Next it is investigated whether the polarity and charge transfer of reaction has been altered or not. For this purpose, the global reactivity indices computed from energies of frontier orbitals are provided in Table 9.
Table 9
Global reactivity indices for reaction B (scheme 2) at M06-2x 6311++(d,p) level
|
Chemical Species
|
η
|
S
|
µ
|
ω
|
N
|
|
2b
|
3.89
|
0.26
|
-6.44
|
5.33
|
0.57
|
|
TS1b
|
1.49
|
0.67
|
-5.08
|
8.62
|
4.32
|
|
3b
|
2.69
|
0.37
|
-4.19
|
3.27
|
4.01
|
From the Table 9 and Table 4, it can be concluded that the reaction is polar in nature and charge is being transferred from diene to dienophile.
MESP of chloro maleic anhydride reveals that electron density on C2 is increased to some extent, as this region is now green instead of blue. Analysis of natural charges indicate that chlorine caused the negative charges of oxygen atoms to reduce and negative charge of C1 to increase. This increase in negative potential of C1 and C2 could also be the reason behind increased barrier for the reaction because in such situation chances of attack of nucleophile gets decreased.
Fukui indices are computed for chloro maleic anhydride, in order to see weather, the most reactive sites have been changed or not.
Table 10
Condensed to atom Fukui (f+) functions for chloro maleic anhydride (2b)
|
Atom
|
C1
|
C2
|
C3
|
C5
|
O4
|
|
f+
|
0.176
|
0.14
|
0.06
|
0.069
|
0.032
|
According to values provided in Table 10, still the most suitable carbons for the attack of nucleophile are C1 and C2. Their ability to get attacked by diene is although somewhat reduced which could be due to electron withdrawing nature of chlorine. This is in consistent with the results of MESP and natural atomic charges.
For knowing the effects of electron donating group on this particular reaction, mechanism of citraconic anhydride with 7H-benzo[a]phenalene is determined. Stability of maleic anhydride is increased on the substitution of methyl group but this time it is stabilized by just 39 Hartree. From Table 11 it can be seen that rise in activation barrier in this case is much higher. This increase in stability cannot justify the increase in activation barrier because increase in stability of chloro maleic anhydride was more than citraconic anhydride but in case of former rise in activation energy was lower.
Table 11
Thermodynamic parameters for reaction C (scheme 3) at M06-2x++(d,p) level
|
Structure
|
ΔH
(kcal/mol)
|
ΔS
(Calmol−1K−1)
|
ΔG
(kcal/mol)
|
ΔE
(kcal/mol)
|
|
1a+2c
|
0
|
0
|
0
|
0
|
|
TS1c
|
70.13
|
-50.16
|
85.08
|
70.6
|
|
3c
|
15.24
|
-53.80
|
31.28
|
12.9
|
Comparison of Fig. 5 and Fig. 8 reveals that in citraconic anhydride energy of LUMO is higher as compared to maleic anhydride thus the gap of LUMOdienopile and HOMOdiene has been increased to 4.8eV, it has made the interactions of these frontier orbitals difficult. Hence, activation energy for the reaction is enhanced to such a large value.
Table 12
Global reactivity indices for structures in scheme 3 at M06-2X 6311++(d,p) level
|
Chemical Species
|
η
|
S
|
µ
|
ω
|
N
|
|
2c
|
4.11
|
0.24
|
-6.18
|
4.65
|
0.61
|
|
TS1c
|
1.31
|
0.76
|
-4.84
|
8.94
|
4.75
|
|
3c
|
2.70
|
0.37
|
-4.10
|
3.11
|
4.10
|
Hardness of methyl maleic anhydride is higher than the hardness of both chloro maleic anhydride and maleic anhydride, which may also be linked to highest barrier of the reaction C (scheme 3). Its electrophilicity index is lower than those of other two anhydrides. As discussed before the reaction in which electrophile has higher value of electrophilicity index are more polar. This shows that this reaction is less polar as compared to other two reactions.
Fukui functions computed for citraconic anhydride are provided in Table 13. Just like the other two anhydrides, C1 and C2 are the carbons with highest probability of attack by diene.
Table 13
Condensed to atom Fukui (f+) functions for citraconic anhydride (2c)
|
Atom
|
C1
|
C2
|
C3
|
C5
|
O4
|
|
f+
|
0.167
|
0.17
|
0.067
|
0.077
|
0.031
|
Methyl group did not cause sufficient increase in negative potential of C2 like chlorine, as evident by the comparison of Fig. 9 and Fig. 7.
Polarity of reaction can also be rationalized by the calculation of global electron density transfer (GEDT) at transition sate of the reaction. GEDT at the transition states of cycloaddition step for all reactions are computed by using the following relation [79]:
GEDT=-∑qA
qA is the charge on atoms of diene fragment of transition state. According to GEDT, reaction of methyl maleic anhydride is least polar. This is in consistent with the results of electrophilicity index.
{kind=link}
{kind=link}