The absorption spectra of the pyrene derivatives show almost identical spectral characteristics in six solvents with different polarities, with slight peak shifts and broadening (Figures 1(b) and S1). While the absorption spectra of Pyr1 and Pyr3 in hexane are very similar, those of Pyr2 and Pyr4 in hexane, while similar, show somewhat redshifted peaks. On the other hand, the absorption spectra are redshifted in the order: Pyr1 > Pyr2 > Pyr3 > Pyr4 in DMSO (Figure 1(d). This relationship between absorption spectral features and solvent polarity suggests that these pyrene derivatives exhibit different degrees of CT character in the ground state. To analysis this in more detail, we acquired fluorescence spectra of these pyrene derivatives, which show that fluorescence emissions are increasingly redshifted and broadened with increasing solvent polarity (Figures 1(c) and S2). As was observed for the absorption spectra, the fluorescence spectra of Pyr1 and Pyr3 in hexane are similar, while those of Pyr2 and Pyr4 in hexane have cognate spectra with somewhat redshifted peaks compared to those of Pyr1 and Pyr3. On the other hand, the fluorescence spectra of Pyr1 and Pyr2 show similar features in DMSO, while those of Pyr3 and Pyr4 have analogous spectra with somewhat blue-shifted peaks compared to those of Pyr1 and Pyr2 (Figure 1(e)). In other words, in the steady state results, Pyr1 (Pyr2) and Pyr3 (Pyr4) show analogous spectral features in the non-polar solvent, while Pyr1 (Pyr3) and Pyr2 (Pyr4) show spectral similarities in the polar solvent, which illustrates that the local excited (LE) states (in hexane) of these pyrene derivatives are affected by the t-butyl substituents, while their CT states (in DMSO) are mainly perturbed by the ortho/meta substituents on the N,N-di(biphenyl) group. The different steady-state trends observed in the non-polar and polar solvents imply that the molecular structures that might undergo TICT are solvent-polarity dependent. Furthermore, the ortho/meta substituents on the N,N-di(biphenyl) group and the t-butyl substituents significantly affect structural change in the excited state.
To explore TICT in these pyrene derivatives, we calculated their ground- and excited-state molecular structures at the B3LYP/6-31G(d) (DFT) level of theory (Figure 2). The dihedral angles between the pyrene moieties and the N,N-di(biphenyl) substituents in these pyrene derivatives were all calculated to be around 50-70° in the ground state, while those in the excited state are close to 90°. The orthogonal geometry endows the molecule with more distinct charge separation between the pyrene moiety and the substituent.26 Moreover, each HOMO is delocalized across the pyrene moiety and the N,N-di(biphenyl) substituent, while the LUMO is localized on the pyrene moiety, which facilitates CT from the N,N-di(biphenyl) substituent to the pyrene moiety (Figures S3-S7). Consequently, it is clear that these pyrene derivatives exhibit CT character accompanied by structural changes in their excited states (TICT), which is consistent with previous results that show that 4-N,N-dimethylaminobenzonitrile is planar in the ground state but has an orthogonal excited-state structure due to its TICT character.26,27
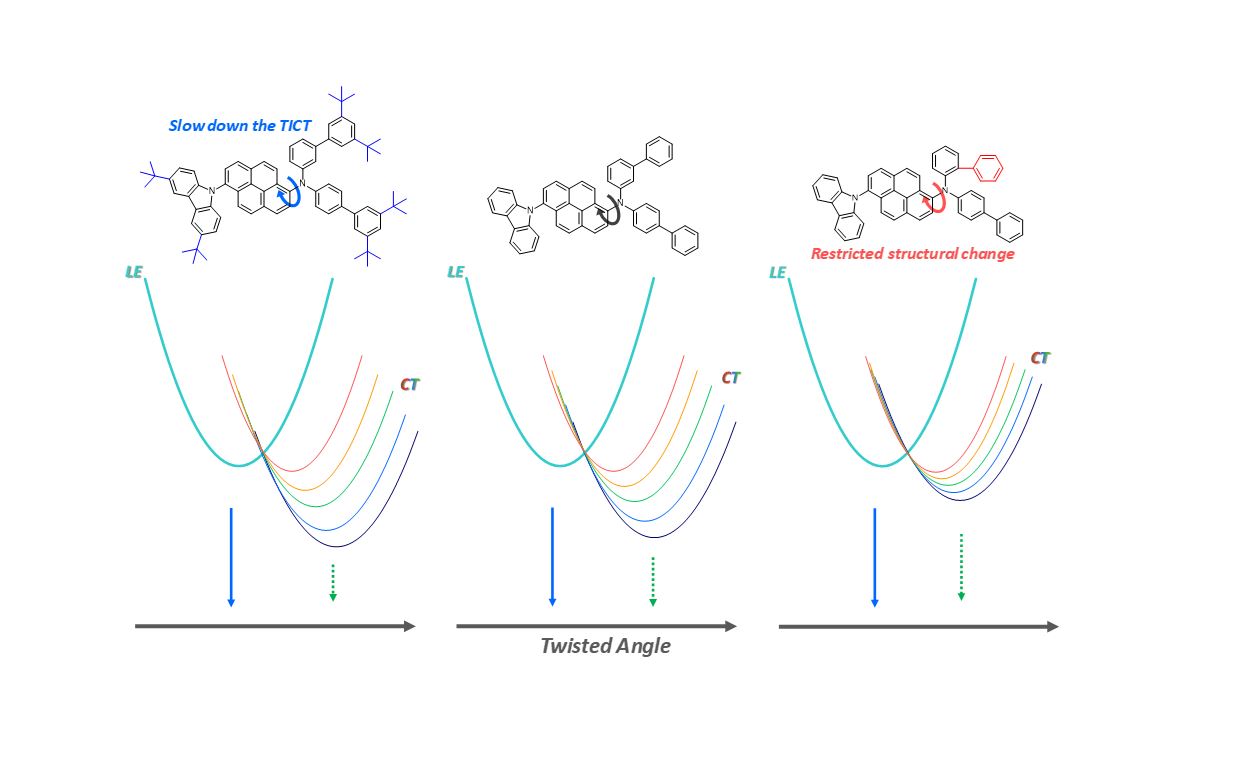
We used the Lippert–Mataga correlation, which provides information on the differences in the dipole moments of ground and excited states, to analysis more accurately on the CT states of our pyrene derivatives (Figure 3a).28,29 The slopes of the Lippert–Mataga plots increase in the order: Pyr4 < Pyr3 < Pyr2 < Pyr1; Pyr1 (Pyr3) and Pyr2 (Pyr4) have almost identical slopes, while those of Pyr1 (Pyr2) and Pyr3 (Pyr4) are different, which indicates that the slope is significantly more affected by the ortho/meta substituents on the biphenyl rings of the N,N-di(biphenyl) moiety than the t-butyl substituents because the ortho/meta substituents severely hinder the ability to undergo structural change, which destabilizes the CT states. These results are supported by the calculated structures of the pyrene derivatives in their ground and excited states. The dihedral angles between the pyrene units and the N,N-di(biphenyl) groups in Pyr1 and Pyr2 are much closer to 90° than those in Pyr3 and Pyr4 (Figure 2), which implies that the CT states of Pyr1 and Pyr2 are better stabilized than those of Pyr3 and Pyr4 because the orthogonal structure enhances charge separation by reducing orbital overlap between the pyrene moieties and the N,N-di(biphenyl) groups.26,27 In other words, the more-orthogonal excited-state structures of Pyr1 and Pyr2 stabilize the CT states and reduced their energy levels, in agreement with the lower slopes observed in the Lippert–Mataga plots of Pyr3 and Pyr4 compared to Pyr1 and Pyr2. Furthermore, these results also suggest that the ortho positions of the phenyl groups of the biphenyl substituent restricts structural change, while structural change is largely unaffected by the t-butyl substituents.
To elucidate details of the TICT mechanism operating in these pyrene derivatives, we measured their fluorescence quantum yields and lifetimes, which were observed to increase with increasing solvent polarity (Figures 3 and S8). Although longer fluorescence lifetimes are generally observed in polar solvents, higher quantum yields in polar media are uncommon because ICT processes typically increase non-radiative rates, resulting in lower quantum yields. In order to diagnose the lower quantum yields observed in nonpolar solvents, we determined the radiative and non-radiative rates of the pyrene derivatives (Figure 4 and Figure S9). The radiative rates tended to be lower in polar solvents (Figure S9), which is in good agreement with previous results.25,30−32 The non-radiative rates, however, were also lower in polar solvents, which is not generally observed for ICT processes.25,30−32 We exclude the possibility of dual emission (emission from both the LE and CT states) because the time-resolved emission spectra (TRES) of the pyrene derivatives show only single components that correspond to their fluorescence spectra (Figure S10).
We used transient absorption (TA) spectroscopy to rationalize the lower non-radiative rates in polar solvents. The TA spectra of the pyrene derivatives in hexane exhibit no spectral shifts in our experimental time window, while those in DMSO show significant blue-shift of the photo-induced absorption with increasing time (Figures 4(a,b), S11–S16). Considering the slow dynamics for CT formation (sub 10 ps) and the blue shifts of the induced absorption at the final time, we associate the fast components of the pyrene derivatives that are somewhat slower in polar solvents to CT formation accompanying structural change.12,17,18 Larger spectral shifts from the initial state to the final state were observed in polar solvents, which indicates that the CT states are better stabilized in polar solvents (Figures S11-S28). The TICT-based excited-state absorption shifts of Pyr1 and Pyr3 in the TA spectra are nearly identical to those of Pyr2 and Pyr4, respectively, which indicates that the CT states can be controlled by the substituents at the ortho/meta positions of the phenyl substituents on the N,N-di(biphenyl) group rather than the t-butyl substituents (Figure S29). A longer lifetime can be induced by a structural change that stabilizes the CT state in a polar solvent. The TICT rates determined from the TA data exhibit similar trends to the corresponding non-radiative rates (Figure 4(c)). In other words, the non-radiative and TICT rates of the pyrene derivatives decrease with increasing solvent polarity, which indicates that TICT is the dominant process that determines the non-radiative rate and significantly affects the fluorescence quantum yield. Consequentially, the slow TICT process associated with a stabilized CT state slows the non-radiative process, which enhances the fluorescence quantum yield. Furthermore, the CT states of the pyrene derivatives are mainly stabilized by molecular torsional motion, which is much slower than a typical charge-separation process. This trend is opposite to that observed for general molecules that fluorescence less in more polar solvents.25,30−32
The TICT rates were analyzed in more detail by considering molecular structure. The pyrene derivatives in toluene, THF, and dichloromethane showed lower TICT rates (Pyr1 > Pyr2 > Pyr3 > Pyr4; Figures 4(c) and S11-S28), while this trend is different in high-polarity solvents (benzonitrile and DMSO: Pyr1 > Pyr3 > Pyr2 > Pyr4), which indicates that insertion of the t-butyl group as well as the ortho substituents on the N,N-di(biphenyl) group reduces the rate of structural change during TICT in a polar solvent.33 Because much smaller structural changes are required to stabilize the CT states of the pyrene derivatives in non-polar solvents than in polar solvents, the effects of structural hindrance and the bulkiness imparted by t-butyl-group insertion and ortho substitution on the excited states are negligible in non-polar solvents. Conversely, the CT states of the pyrene derivatives are stabilized in polar solvent through large structural changes; consequently, ortho substituents on the N,N-di(biphenyl) group and the t-butyl-group insertion significantly affects their CT states. In particular, the t-butyl substituents affect the TICT rates more than the ortho substituent, which implies that steric hindrance affects the TICT rate less than the bulkiness of the molecular structure. We rationalize this in terms of the bulkiness of the t-butyl substituents that increase the volume of the rotational groups in the TICT process, which leads to the slow TICT rates observed for Pyr2 and Pyr4. However, the steric hindrance of the N,N-di(biphenyl) group affects the TICT rate somewhat less, although it significantly destabilizes the CT state by hindering rotation. In other words, the CT rates are significantly affected by the bulkiness (volume) of the molecules rather than the steric hindrance associated with rotating groups, while the energy levels of the CT states are mainly affected by the steric hindrance of the rotating groups. To sum up, TICT processes in non-polar solvents are negligibly associated with structural change and are not significantly affected by modifications to the molecular structures. On the other hand, the TICT rates in polar solvents are controlled by the bulkiness of the t-butyl groups in these pyrene derivatives because structural changes are significantly affected by molecular volume. In addition, ortho/meta substituents on the N,N-di(biphenyl) groups of these pyrene derivatives negligibly affect CT rates because they hardly affect molecular volume; rather they stabilize the energy levels of the CT states.
{kind=link}