Triple-negative breast cancer remains the most challenging breast cancer subtype to treat. Recently, targeted therapies for TNBC treatment have been quested. PI3K/AKT/mTOR pathway, RAS/MAPK pathway, EGFR, JAK/STAT pathway, and NOTCH are some potential targets in clinical trials (27). However, therapies directed to specific molecular targets have rarely achieved effective improvement during clinical remedy, and chemotherapy remains the standard of care. Nonetheless, over 30% of TNBC patients become resistant to chemotherapy and it lead to the failure of treatment (4). Drug efflux (overexpreesion of ATP binding cassette transporters proteins, and P-glycoprotein) (28), up- or down-regulated autophagy flux, de-regulation of distinct cell intrinsic processes (the nuclear proto-oncogene c-MYC)(29), growth factor signaling, and DNA repair have previously been explored as the mechanisms of TNBC chemo-resistance. Therefore, there is still an urgent need to develop novel molecular targets to improve the therapeutic benefit of chemotherapy given in first-line settings, including the patients with advanced, chemotherapy resistant TNBC.
Autophagy has been recognized as an effective cancer escape mechanism and the reason for drug resistance in multiple cancer types. Early clinical trials have demonstrated the feasibility and potential benefit of inhibiting autophagy in multiple cancer types, including pancreatic cancer, lung cancer, melanoma, and multiple myeloma (30). Studies have demonstrated that autophagy can be inhibited pharmacologically by targeting the different stages of the autophagic process. Early-stage inhibitors like 3-MA and LY294002, function on class III phosphatidylinositol 3-kinase (PIK3C3) and block the formation of autophagosomes (31, 32). The inhibitors that target a late stage of autophagy like chloroquine, BAF A1, leupeptin, etc. impair the fusion of the autophagosome with lysosome or hydrolyzed function of lysosome. CQ/HCQ is currently the only clinically available drug to inhibit autophagy. However, the high effective concentration of CQ may cause an off-target effect. Therefore, it is necessary to discover more potent autophagy regulators with the potential to develop into novel anti-tumor drugs.
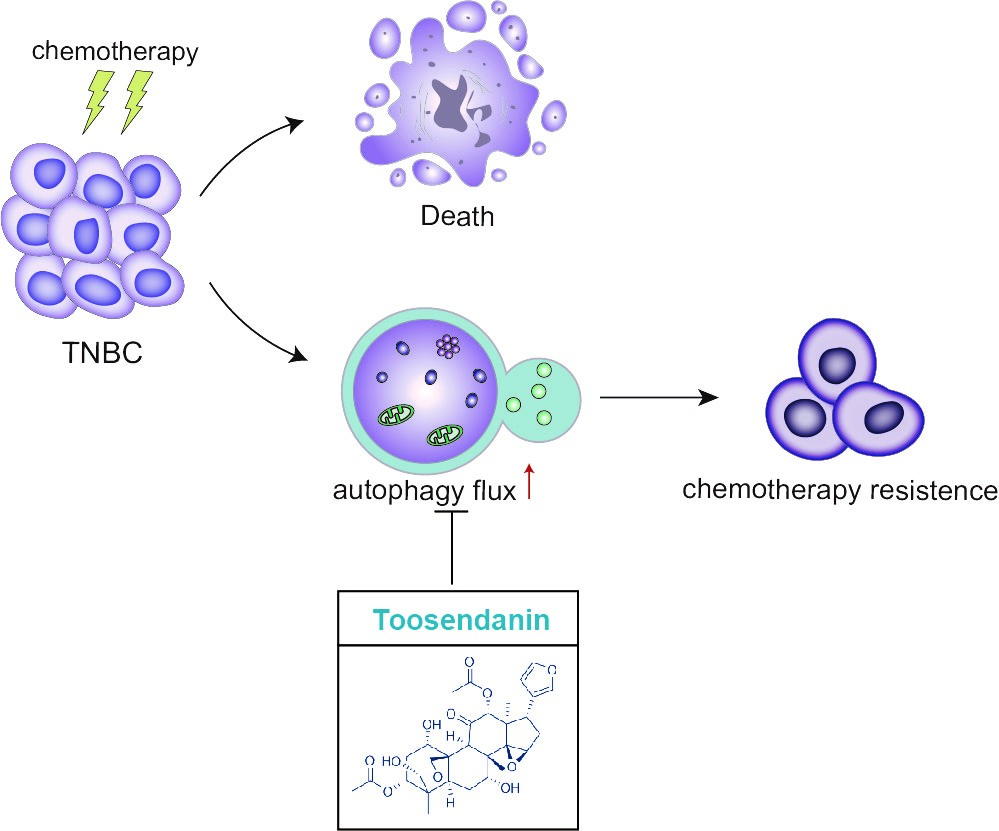
Toosendanin (TSN), a triterpenoid extracted from Melia toosendan Sieb. et Zucc, has been reported to possess antioxidant, anti-inflammatory, and anti-allergic activities (33, 34). Recent studies have revealed potential anti-cancer activity of TSN in diverse cancer models, such as glioblastoma, Ewing's sarcoma, gastric cancer and hepatocellular carcinoma (15, 16, 33, 35, 36). Luo W et al. (33) reported that TSN could inhibit transforming growth factor-β1-induced epithelial-mesenchymal transition and migration through ERK/Snail pathway in lung cancer cells. Recently, Wang H et al. provided evidence that TSN -induced apoptosis is associated with the κ-opioid receptor/β-catenin signaling axis in colorectal cancer cells (37). Remarkably, TSN has been reported to mediate cisplatin sensitization through down-regulation of Annexin A4 in lung cancer(38). These studies provided a possible mechanistic explanation for the anti-tumor effect of TSN. To date, only one study described the relationship between TSN and autophagy in mammal cells. However, this study claimed that TSN developed an apoptosis-sensitizing effect by inducing autophagy in lung cancer cells (39). When checking their data, no autophagy flux assay was applied in the study thus their interpretation of the results could be problematic. In our study, we provided a careful characterization of the effect of TSN on autophagy according to Autophagy Modulator Scoring System (AMSS) (40), and confirmed that TSN is a late-stage autophagy inhibitor. Considering the high potency of TSN in inhibiting autophagy both in vitro and in vivo, we believe that autophagy inhibiting activity is an important anti-cancer mechanism of TSN.
TSN has been used in clinics for treating intestinal ascariasis in China (41). However, liver toxicity of TSN in mice has been reported (42), raising the concern for the use of TSN as an anti-cancer drug. According to the mice data, TSN did not induce obvious liver toxicity at the dosage up to 40mg/kg (intragastric administration), implying there is still a safety window for the clinic use of TSN. Due to different drug administration routes, we cannot directly compare the dosage we used in the current study (0.5mg/kg, intraperitoneal administration) with that used in the previous study (40mg/kg, intragastric administration). However, our data showed that at the dosage sufficient to inhibit autophagy in tumor tissue, TSN did cause obvious liver toxicity according to the liver index. Nevertheless, extensive safety characterization is still need to evaluate the safety of TSN for long-term application.
Up-regulation of autophagy observed in tumor cells following anti-cancer treatment is regarded as a protective response (43–45), and therapeutic targeting of autophagosome formation/fusion might represent a novel molecular avenue to reduce the emergence of chemo-resistance (5). Irinotecan was initially approved for the first-line treatment of metastatic colorectal cancer and later was also approved for lung cancer treatment (17). Irinotecan can be suitable for TNBC treatment given the fact that a considerable portion of TNBC tumors harbor mutations in genes required for DNA repairment (18). Indeed, recent studies highlighted the potential of irinotecan in the BRACness TNBC treatment (18–20). In this report, we showed that irinotecan/SN-38 induced autophagy in TNBC cells as a survival mechanism, and autophagy inhibition by TSN sensitized TNBC cells to irinotecan/SN-38 chemotherapy. Taken together, the data generated from this study reveals a novel therapeutic strategy for TNBC treatment by combination of topoisomerase I inhibitor and autophagy inhibitor.
{kind=link}