In late July, a 32-year-old male with mild flu-like symptoms of fever, headache, cough, fatigue, and sore throat was referred to the Santo Antônio de Pádua municipal health department in the northeastern region of the state of Rio de Janeiro, Brazil. After two days from the onset of symptoms, the diagnosis of COVID-19 was confirmed by SARS-CoV-2 reverse transcriptase polymerase chain reaction (RT-PCR) assay of a nasopharyngeal swab. Given the mild symptoms, no hospitalization was required and the patient was quarantined at home. The patient did not report any comorbidities and had not been vaccinated by the time the sample was collected.
Whole-genome sequencing analysis detected 73 intra-host single nucleotide variants (iSNVs) with allele frequency > 5% and depth > 100 reads. Of these, 26 were lineage-defining mutations exclusively found in the variant of concern (VOC) Gamma (P.1) and 32 were from Delta (AY.33; Table S1). By using a hypergeometric distribution approach1, we estimate an overall haplotype frequency of approximately 16% and 82% for Delta and Gamma, respectively. Nevertheless, two iSNVs characteristic of VOC Delta (ORF1ab: I1091V - AF = 94%; and ORF7b: T40I - AF = 70%) were found with a high frequency in our analysis, suggesting that some genomic copies from Gamma haplotype could also harbor the same mutations (Table S1).
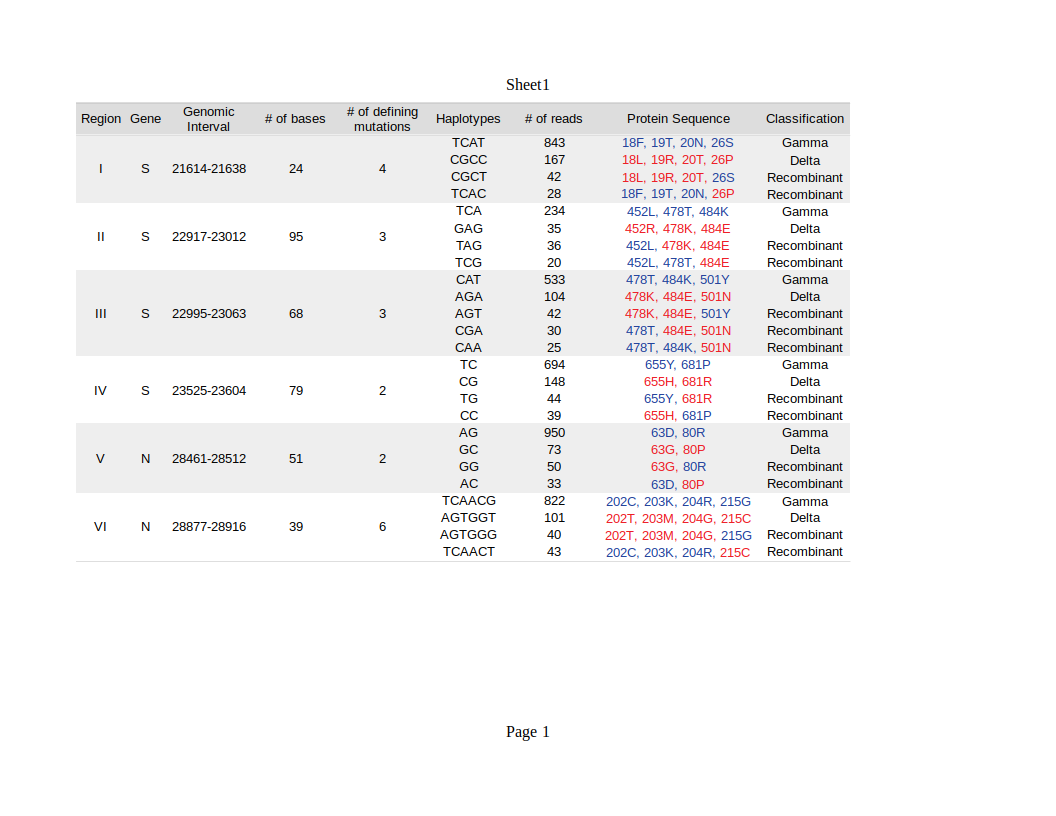
We then sought to investigate the occurrence of within-host recombination events between the two lineages using next-generation sequencing. In theory, mutations characteristic of Gamma should be kept in different reads from those defining mutations of Delta. We divided the SARS-CoV-2 genome into windows with at least 100 nucleotides, which is smaller than the read length of the Illumina COVIDSeq Test (Illumina) sequencing kit used in this study. Next, we only selected windows that intersected at least two iSNVs, being one lineage-defining mutation from Gamma and the other from Delta. We aimed to detect unfragmented reads that covered the entire window harboring the iSNVs selected. A total of six recombinant candidate regions across the SARS-CoV-2 genome were found (see Supplementary Material), four mapped in the spike gene and two in the nucleocapsid gene (Table 1). We detected an average of two recombinant haplotypes supported by reads harboring a combination of the discriminating mutations from Gamma and Delta (Table 1; Figures S1-S6). In addition, the within-host recombinant sequences were formed by a single breakpoint event with a minimal and maximum interval of 15 and 75 nucleotides between two discriminate mutations where an event of recombination was observed, respectively.
Although the number of reads intersecting the discriminating iSNVs in the recombinant haplotypes seems to be small, we also observed a huge number of reads covering each mutation separately (Table S1; median read depth: 1947). Thus, the read coverage and minor allele frequency in each site suggest that these mutations were not caused by low mapping quality or miscalling variant issues. The hypothesis that the recombination might be the product of laboratory contamination is highly discouraged due to the protocol used by us. All viral particles are inactivated before RNA extraction, which stops replication and, consequently, impairs recombination from happening due to sample contamination. In addition, the high efficiency of ARTIC pool of primers utilized in our library construction rule out the possibility of the recombinant fragment being made by the initial PCR amplification step. Moreover, the 100 nucleotide window used in our genome scan and the similarity of sequence breakpoints analyzed in different genome regions make it difficult to imagine that it can be produced by the PCR step used in our library construction.
Recent studies reported the detection of SARS-CoV-2 recombinant lineages circulating at a low frequency2–6. Nevertheless, these studies were restricted to detecting the inter-host dissemination of genomes post-recombination events, which could be biased by spurious mutations generated due to library preparation, laboratory contamination, and unavailability of raw sequenced data. Here, we report the first case of intra-host SARS-CoV-2 recombination during a coinfection by the VOC Delta (AY.33) and Gamma (P.1) supported by sequencing reads harboring a mosaic of lineage-defining mutations as well as putative sequences breakpoints. We did not detect the circulation of recombinant sequences in the state, so far. Finally, Gamma and Delta variants were notable by causing an elevated number of Covid-19 cases and deaths in Brazil and worldwide, respectively. Both VOC are known to enhance SARS-CoV-2 transmissibility and induce an immune-scape response. Therefore, recombination between both sequences could represent the emergence of novel variants of concern with new phenotypic combinations.
{kind=link}