3.1 Isolate selection for genomic and metabolomic analysis
All isolates under investigation were initially identified morphologically and then confirmed as F. poae by TEF1-α gene sequence homology with other sequenced Fusaria at the Fusarium-ID website (http://isolate.fusariumdb.org/blast.php)(24). From the original selection of 184 F. poae isolates collected from FHB symptomatic wheat, barley and oat heads from 2006 to 2017, a subset of 38 isolates were selected for detailed genomic and metabolomic analysis. Additional File 1 contains a list of all isolates screened. The selection of the 38 isolates was based on: genetic variance associated with TEF1-α and trichothecene biosynthetic genes TRI1 and TRI8 (inferred phylogenetic tree is in Additional File 2); diverse metabolomic signatures from ultra-high performance liquid chromatography coupled to high resolution mass spectrometry (UPLC-HRMS) data of extracts from isolates grown on YES media; and variation in host crop and geographical origin. TRI1 and TRI8 were chosen due to previous analyses which showed variations in these genes led to alternate trichothecene modifications(25–27) and higher sequence divergence observed within previously genome sequenced F. poae isolates(17).
Genome assembly using Illumina sequence data of the 38 chosen isolates produced a median of 1375 scaffolds per genome and a mean genome length of 39.25 Mb. A summary of Illumina genome assembly statistics can be found in Additional File 3. To evaluate the completeness of the assemblies we identified BUSCO (Benchmarked Universal Single Copy Orthologs)(28) gene analysis using the Hypocreales_odb10 database, which revealed all genomes were over 97% complete, with the exception of one poor quality genome (Fp030; 80.7%), and one failed sequencing run (Fp029, not included in genomic analysis).
3.2 A high-quality genome for Fp157 confirmed the secondary metabolite biosynthetic potential in F. poae and the presence of accessory chromosome-associated sequences
F. poae isolate Fp157 was selected for long-read sequencing using the Oxford Nanopore platform (340,123 filtered reads, mean length of 21,465 bp and mean qscore of 10.24). We generated a high quality genome (Figure 1) with four core chromosomes exhibiting strong macrosynteny to the previously genome sequenced Belgian F. poae isolate 2516(17) as well as F. graminearum isolate PH1(29). Additional File 4 contains genome assembly statistics for Fp157, and Additional File 5 contains LASTZ(30) dot plots comparing core chromosome synteny between Fp157, F. poae 2516 and F. graminearum PH1. A total of 14,114 genes were predicted, with two additional genes manually annotated based on blastn matches to biosynthetic genes (FpPKS2 and FpNRPS4, discussed in text). Core chromosomes Chr1 and Chr2 represent ‘telomere to telomere’ sequences with putative centromeric regions and telomeres comparable to the length, position and GC content of those described from F. graminearum PH1(29). Centromeres consist of approximately 50Kb-long regions averaging 15% GC content, and telomeres show canonical ‘TTAGGG’ repeats followed by a 1500bp region averaging 37% GC content. Chr4 has telomeric repeats at the 5’ end, whereas the 3’ end encodes predicted rDNA repeats, also congruent with F. graminearum PH1. Chr3 is missing a telomeric sequence at the 5’ end and when compared to isolate 2516, subtelomeric regions appear inverted and rearranged. Lastly, Chr1 shows an approximately 1Mb inversion compared to its counterpart in isolate 2516.
In addition to the core chromosomes, 9 contigs were assembled in the Fp157 genome ranging in size from 100,057bp to 1,877,593bp. Contig_5 is 140,862 bp long, representing the mitochondrial genome, and was not annotated. Contig_8 is 122,757bp of rDNA repeats and is virtually identical to the 60Kb of rDNA repeats associated with the 3’ end of Chr4. The remaining seven contigs cumulatively total 5,205,433bp and are presumed to represent AC sequences based on a number of factors. First, predicted ACs have genetic content that is congruent with the published content of predicted ACs in Belgian F. poae isolate 2516(17). Second, there is a lack of macrosynteny to sister species F. graminearum PH1 core chromosome sequences. Third, contigs are predicted to be less affected by repeat-induced point mutations (RIP, predicted by sliding-window dinucleotide frequency analysis(31)), when compared to core chromosomes, a pattern also congruent with F. poae 2516 ACs. Fourth, the contigs show elevated repetitive element content compared to core chromosomes, as seen in many confirmed fungal ACs(32). Finally, the contigs show lower predicted gene densities when compared to core chromosomes, another characteristic feature of ACs(20,33) (Figure 1).
Within the AC-associated contigs, there are three putative centromeric regions identified by similar size and GC content to core centromeres (contig_1, 52.7 kb averaging 14.5% GC content; contig_3, 53.3 kb averaging 14.7% GC content; contig_4, 35.2 kb terminating at end of contig, averaging 15.6% GC content). Additionally, six sets of telomeric repeats were identified and are all located on contig terminal regions. We therefore suggest there are three ACs in total, however further experimental verification including electrophoretic karyotyping is needed to confirm the number and size of the ACs, and to build telomere-to-telomere assemblies of their contents.
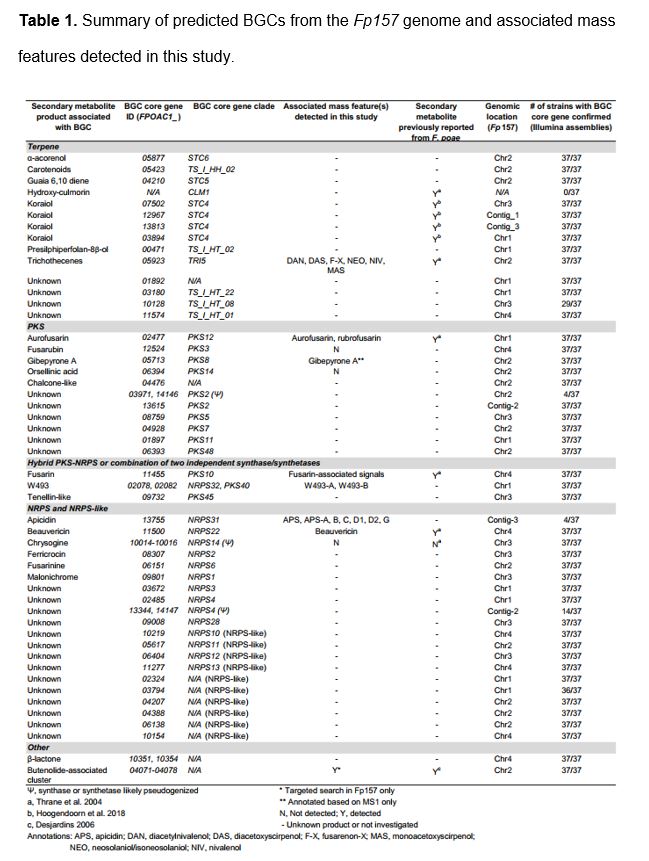
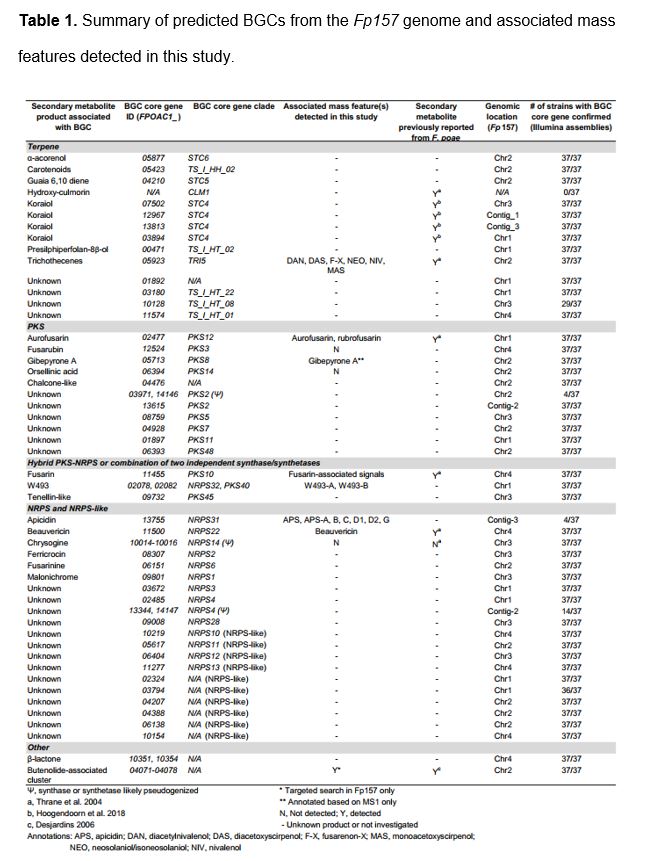
AntiSMASH 5.1(34) analysis of the Fp157 genome predicted 43 discrete BGCs which encoded various core scaffold genes including 12 polyketide synthases (PKS), 13 terpene synthases, 10 non-ribosomal peptide synthetases (NRPS), 10 NRPS-like synthetases (usually a single NRPS module lacking a canonical domain) and 2 hybrid PKS-NRPS genes (Table 1). One cluster was predicted to be involved in the formation of β-lactones. Blastx comparison of NRPS adenylation domains, PKS ketosynthase domains and full-length PKS genes (all domains) to published databases of Fusarium-associated PKS and NRPS genes(15,35) indicated all PKS and NRPS genes are orthologs of genes previously associated with Fusaria, and approximately half are associated with known products.
3.3 Chemical phenotyping of F. poae isolates
Chemical phenotypes for each of the 38 isolates grown in vitro were generated to visualize patterns in mass feature detection frequencies, and untargeted mass feature diversity. Mass features were obtained from UPLC-HRMS profiles of extracts from isolates grown on five media conditions. Each media condition was chosen to diversify sources of nitrogen, sugars, salt stress and starvation stress. Media formulations used are detailed in Additional File 6. Mass feature intensities were converted to binary presence/absence for each media condition, and then averaged across all media conditions into a consensus phenotype for each isolate. Figure 2 represents consensus chemical phenotypes from all isolates and includes all mass features discussed in this study as well as a subset of unannotated signals (see Additional File 7 for expanded analysis).
Hierarchical cluster analysis grouped the consensus chemical phenotypes (‘metabolomes’) by metabolite detection pattern similarities between isolates and between mass features (Figure 2, left and top dendrograms). Mass features annotated as trichothecenes and fusarins were present in large clusters due to the many functional alterations present in these molecular families. Close examination of raw MS data from isolates Fp016 and Fp038 revealed extracts from all media conditions were dominated by the relative abundance of fusarin-associated signals. This likely led to a skewed mass feature profile (with many mass features falling below the limit of detection) due to sample dilution prior to injection, and possibly again during data preprocessing, which could explain the absence of beauvericin and some trichothecene-associated signals from these isolates. Fusarins made up a significant portion of signals from nearly all isolates on all media types, with the exception of isolates Fp059, Fp039 and Fp033 which had lower frequencies of fusarin-associated signals relative to the other isolates. We concluded that isolates exhibited varying levels of core-chromosome associated signals. Furthermore, we noted four isolates, Fp157, Fp013, Fp088 and Fp072, exhibited mass features which were absent from all other isolate profiles, and were therefore predicted to be AC-associated based on the presumption that AC biosynthetic content varies across populations of F. poae. These mass features were annotated as apicidins.
3.4 Annotation of type A and B trichothecenes
Comparison of trichothecene-associated signals to commercial standards confirmed the presence of both types A and B which have been previously associated with F. poae. Isolates grown on YES media appeared to produce the most trichothecene-associated mass features, a pattern which is unsurprising since this media is rich in sucrose, a known trigger for trichothecene production in the closely-related species F. graminearum(36). Mass features associated with 15-diacetoxyscirpenol, 15-monoacetoxyscirpenol, neosolaniol and fusarenon-X were confirmed by comparison to standards. A feature with the same m/z and similar fragmentation pattern as neosolaniol was detected, eluting slightly later than neosolaniol, and is therefore suggested to be iso-neosolaniol (either 4,8 diacetyl or 8,15 diacetyl form). Additionally, mass features were annotated as diacetylnivalenol and scirpentriol based on in silico fragmentation comparison. A mass feature matching a commercial nivalenol standard was detected from most isolates but was a low-intensity signal compared to other trichothecenes. Other signals associated with trichothecene biosynthesis matched m/z and chemical formulas of trichothecene precursors such as isotrichotriol, or analogs of the major type A and B trichothecenes detailed above, and were tentatively annotated as “trichothecene-associated” due to the absence of publicly available MS2 spectra or commercial standards at this time.
3A-deoxynivalenol, 15A-deoxynivalenol, T-2, HT-2 and T-2 tetraol- associated m/z were not detected from any isolate grown in this study. To test whether isolates had the genetic capability to produce T-2 or HT-2 toxin, we surveyed (by blastp) the 37 F. poae Illumina genomes using the acetyl transferase TRI16 from F. sporotrichioides, shown to facilitate esterification of the C-8 hydroxyl group in trichothecenes during production of T-2 toxin(37), and no TRI16 homologs were detected.
3.5 Confirmation of aurofusarin, beauvericin, fusarin, gibepyrone, W-493 and other metabolites
Next, we annotated mass features associated with non-trichothecene mycotoxins. Beauvericin was confirmed by comparison to a commercial standard. Cyclodepsipeptides W-493 A and W-493 B(38) were annotated by comparison to GNPS-supplied MS2 fragmentation spectra (Mirror plots comparing MS2 spectra are in Additional File 8). Fusarin-associated signals were numerous, likely owing to the multiple stereoisomers commonly observed from fusarin-producers(39,40). Features matching the m/z, predicted in silico fragmentation patterns and UV spectra of aurofusarin and its precursor rubrofusarin were detected from most isolates but production was not consistent to isolate groupings or media conditions. A mass feature matching m/z of gibepyrone A was detected, but could not be confirmed due to lack of standards and experimentally derived MS2 spectral database representation, and in-silico MS2 spectra generation was inconclusive. Additionally, a gene cluster homologous to the butenolide-associated cluster in F. graminearum(41) was detected in the Fp157 genome. We reasoned that due to the highly polar nature of butenolide, it likely eluted with the injection peak during chromatography and was therefore excluded from our UPLC-HRMS chemical phenotypes (the injection peak was sent to waste to prevent soiling of the MS inlet due to media components). Extracts from Fp157 were re-profiled by UPLC-HRMS without diverting the injection peak, and butenolide production was confirmed in this isolate (see Additional File 9 for details).
Several secondary metabolites predicted from the antiSMASH analysis for F. poae (Fp157) were not detected. Notable absences include fusarubin (and other analogs associated with the fsr1 or PKS3 cluster(42)), orsellinic acid and chrysogine. Fusarubin and orsellinic acid analogs may be among the unannotated signals here and will be the subject of further investigation. Closer inspection of the chrysogine-associated NRPS14 homolog in F. poae isolates indicates it has been disrupted by an 800bp insertion of very low GC content (<10%) in all isolates including the Belgian F. poae isolate 2516. Although hydroxy-culmorins have been previously detected from F. poae isolates(9), we were unable to find homologs of the longiborneol synthase gene CLM1, shown to be required for culmorin production in F. graminearum(43), in any of the F. poae assemblies generated in this study. Hydroxy-culmorin-associated mass features were detected in the metabolomes of nearly all isolates (except Fp016), but lack support due to the absence of available commercial standards. The presence of hydroxy-culmorins is thus insufficiently supported at this time to warrant annotation.
Taken together, our data confirms Eastern Canadian F. poae isolates produce similar chemical profiles to European F. poae isolates when grown in vitro, as pertaining to trichothecene, fusarin, beauvericin and aurofusarin production. Our untargeted analysis of chemical profiles detected cyclodepsipeptides W-493-A and B associated signals, and highlighted mass features not matched in our database of known Fusarium mycotoxins which could represent undescribed secondary metabolites (Figure 2).
3.6 The production of apicidin and its derivatives is linked to the accessory chromosome in Fp157
We investigated the detection of apicidins. Mass features were detected primarily from broth extracts of four of the 38 isolates cultured (Fp013, Fp072, Fp146, Fp157), and included features with m/z and isotope ratios matching apicidin (APS) and APS analogs A, B, C, D1, D2, and G(44,45). To confirm these annotations, we used feature-based molecular network analysis and MS2 spectral matching (Figure 3A). Network analysis showed all isolate-specific signals grouped into a single subnetwork. MS2 spectra from three mass features matched publicly available, experimentally-derived MS2 spectra from APS, APS B and APS C (mirror plots in Additional File 10). MS2 data from APS-A, D1, D2 and G, were not represented in experimentally-derived MS2 databases at the time of analysis. However, APS-A, D1 and D2 were supported by in silico predictions of potential fragmentation patterns from known molecular structures using Sirius / CSI Finger-ID. Lastly, APS G was annotated by manual examination of the MS2 spectra (Figure 3B, see Additional File 11 for expanded analysis of APS G signals, and Additional File 12 for expanded network analysis of APS-associated signals). Molecular networking analysis indicated there were at least three unannotated signals which matched fragmentation patterns of the apicidin-like signals, suggesting they are novel APS analogs. The most intense of the three unknown APS-like signals had the same m/z as apicidin D1 [M+H]+ peak (m/z 640.3705) but was determined to be a [M-H2O+H]+ ion by the MZMine IIN module, with the [M+H]+ missing from the raw data, although a [M+Na]+ adduct signal was evident. The other two were detected at relatively low intensities and were not further investigated.
Genomic analysis of the Fp157 isolate revealed homologs of all 11 genes previously shown to be co-regulated during APS production(50), are present in a single, near-contiguous cluster located on AC-associated contig_3. We annotated this cluster as ‘FpAPS’. We compared the FpAPS cluster to the APS cluster annotated from Jin et al. 2010 using blastn (Figure 3C) and determined all genes relating to APS production are found in the same relative order, with the exception of a DNA-transposon insertion between APS8 and APS9 in Fp157. This transposon matches DTF_Fot5-A from a previously published library of transposons described from the Belgian F. poae isolate 2516(17), but appears to be fragmented (Figure 3C).
Additionally, four genes with predicted roles in biosynthesis were found adjacent to the FpAPS cluster in Fp157 and are uniquely present in APS-producing isolate genomes. The co-localization of these genes to the FpAPS cluster indicates they may be involved in APS-associated molecular family diversification. FPOAC1_13756 is a predicted amidase, which appears to have a DTA_Obara DNA transposon insertion (see (51) for explanation of TE nomenclature used here). This gene is also present in the homologous APS cluster in F. sporotrichioides and has not previously been associated with apicidin production. Three additional predicted biosynthetic genes are adjacent to the FpAPS cluster: FPOAC1_13757 is an NRPS-like gene with an AMP-binding domain, FPOAC1_13758 is an O-methyl transferase and FPOAC1_13759 is an oxidoreductase with similarity to the ELFV-dehydratase family. Interestingly, when searched against the NCBI database, all three genes have top blastn hits in a three-gene cluster encoded by the strawberry anthracnose-causing mold, Colletotrichum nymphaeae isolate SA01, which is suggested to grow endophytically in weedy grasses(52). Furthermore, the trio of genes are interspersed by two repetitive sequences with homology to ‘miniature-impala’ or ‘mimp’ TEs which have been associated with pathogenicity genes on Fusarium oxysporum ACs(53). The FpAPS cluster and adjacent genes are present in the genomes of all isolates from which APS-associated mass features were detected.
The possibility of the FpAPS cluster originating by horizontal gene transfer from another species was investigated by performing a Blastn analysis of the FpAPS genes against the NCBI nucleotide database. Top hits for all FpAPS genes matched coding sequences from close relatives F. sporotrichioides and F. langsethiae, with most at ~97% nucleotide identity, are provided in Additional File 13. As assemblies for F. sporotrichioides and F. langsethiae are not currently at chromosome-level, we cannot yet infer whether their APS-like BGCs are on ACs or core chromosomes. However, the location of the contig breaks in the F. langsethiae 201059 genome strongly suggests that it has the same transposable element (TE) insertion site between APS8 and APS9 and therefore this APS-like cluster is likely to be either on an AC or accessory region in this isolate.
Finally, to assess the frequency of occurrence of FpAPS1 in our collection of 193 Ontario and Quebec F. poae isolates as well as 10 international isolates, we designed PCR primers which would specifically amplify a 150 bp nucleotide sequence from F. poae APS1 and screened the genomic DNA (see Additional File 14 for representative sample gel lane). Of these, 15 isolates tested positive for the APS1 gene, including an isolate collected from Ontario wheat in 2006 (DAOMC 239526), indicating the presence of the FpAPS1 has persisted at least as long as the time period under study in Eastern Canada. See Additional File 1 for results of APS1 screening. Active APS production was confirmed from 4 out of the 15 FpAPS1 containing F. poae isolates (as they were the only ones included in the in-depth metabolomics analysis described above). None of the international isolates tested positive for the APS1 gene.
{kind=link}
{kind=link}