sel-12 mutants show decreased autophagy that is rescued by reducing mitochondrial calcium uptake
Like other age-related diseases, AD is associated with a general decline in proteostasis that contributes to the buildup of toxic protein aggregates, leading to neuronal dysfunction and death (27, 28). It has been postulated that initial impairments to proteostasis pathways promote the accumulation and aggregation of misfolded proteins observed in neurodegenerative disorders, including the accumulation of the aggregation-prone Aβ and tau, whose plaque deposition and tangle formation, respectively, are considered the hallmarks of AD (29-33). Utilizing several models of proteotoxicity, we previously reported that sel-12 mutants have a severe defect in proteostasis resulting from elevated ER to mitochondrial calcium signaling (16). Two major systems are responsible for degrading misfolded or damaged proteins and are critical for maintaining integrity of the proteome: the ubiquitin-proteasome system and the autophagy-lysosomal pathway. To investigate the activity of these pathways in sel-12 mutants, we first examined proteasome activity. As a reporter for proteasome function, we analyzed animals expressing ubiquitin (ub(G76V)) tagged to GFP, which is readily degraded by the proteasome in wild type animals, but accumulates if proteasome activity is perturbed (34) (Fig. 1A). Indeed, unlike animals treated with a proteasome inhibitor (bortezomib), animals carrying a sel-12 null allele, sel-12(ty11), showed similarly low fluorescent intensity as wild type animals (Fig. 1A-B), thus, indicating typical proteasomal degradation of the ub(G76V) tagged GFP. These data are consistent with normal proteasome activity of sel-12 mutants previously reported (16).
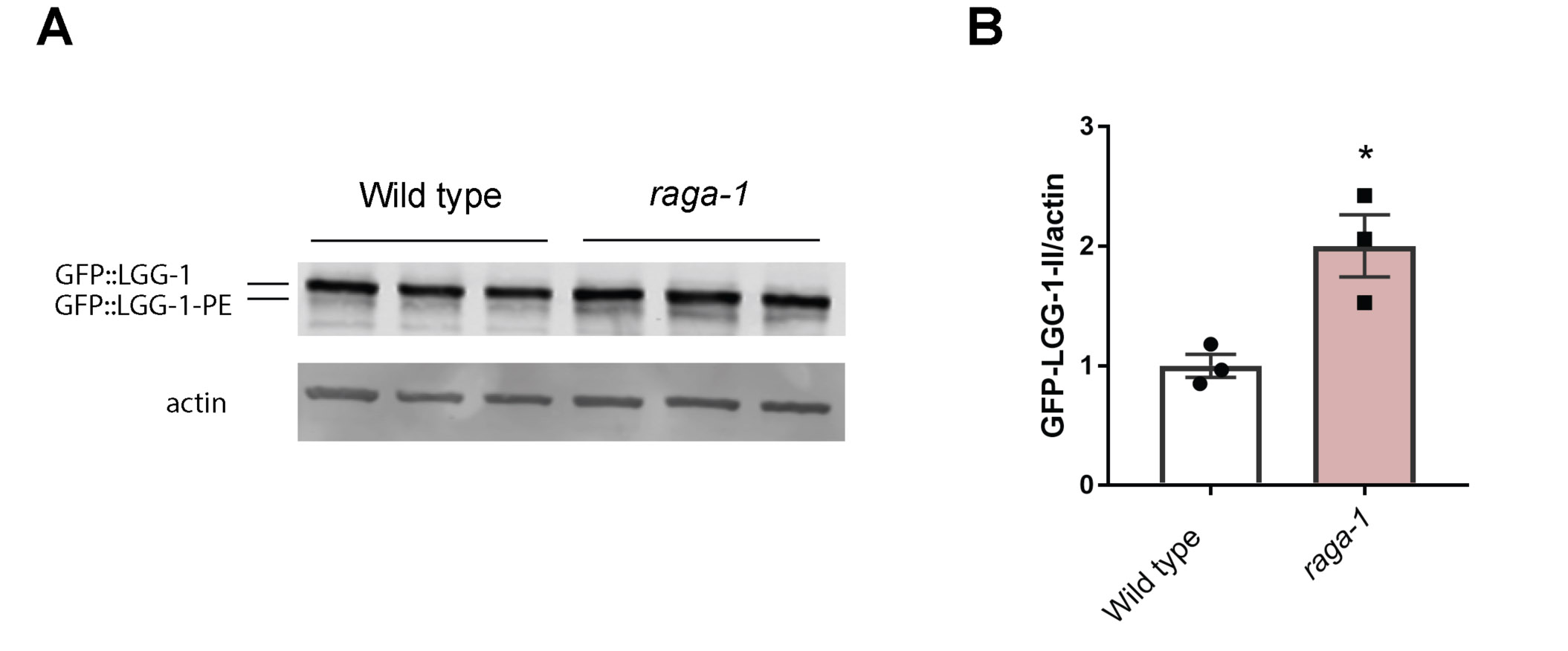
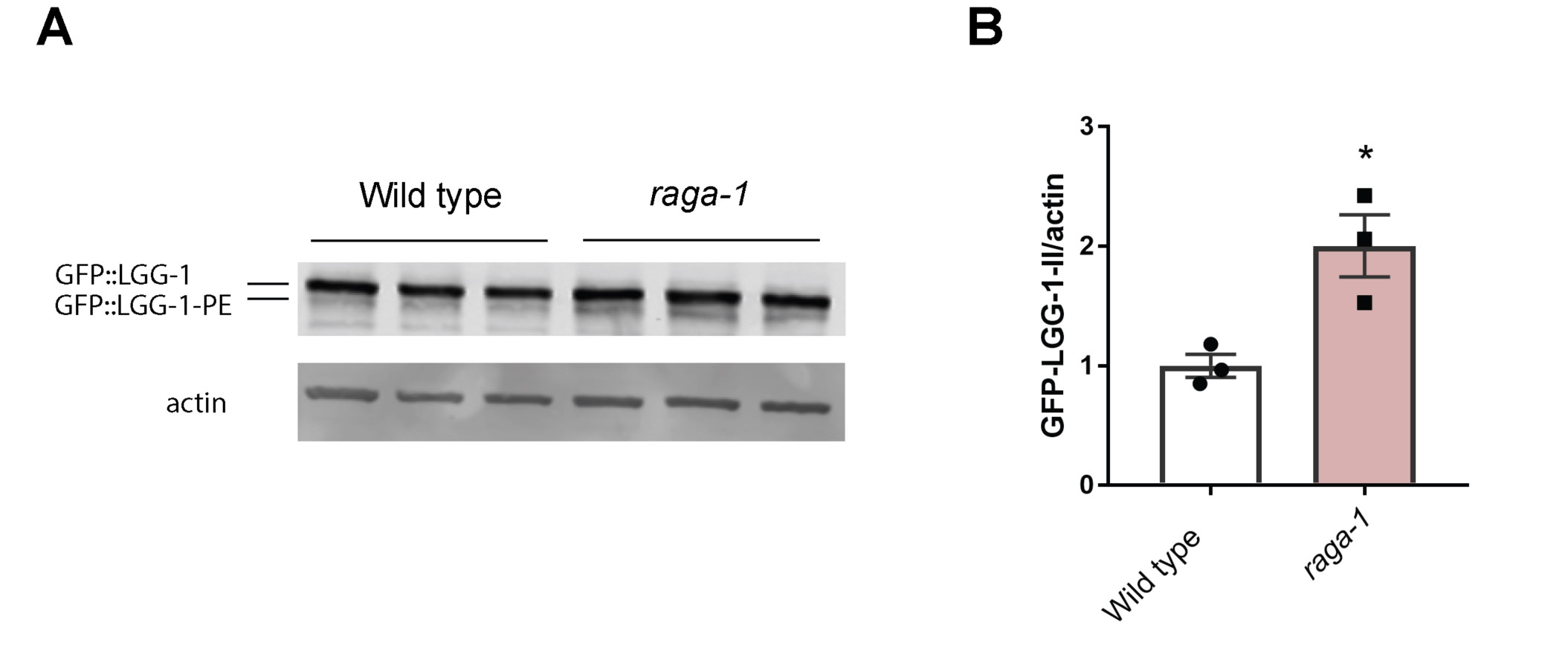
Next to investigate the activity of autophagy-lysosomal pathway in sel-12 mutants, we utilized GFP tagged LGG-1 to visualize autophagosome formation. LGG-1 is the C. elegans ortholog of LC3/Atg8, which is incorporated into and decorates pre-autophagosomal and autophagosomal membranes and organizes into puncta during autophagy and, thus, is a widely used autophagy marker (35-37). Compared to wild type animals, we observed a significant reduction in the number of LGG-1::GFP puncta in the hypodermal seam cells (Fig. 1C-D) and body wall muscle (Fig. 1E-F) of sel-12 mutants. To determine whether elevated ER-mitochondrial calcium signaling in sel-12 mutants is responsible for decreased puncta formation, we introduced into the sel-12 mutant background a null mutation in the mitochondrial calcium uniporter, mcu-1, which reduces mitochondrial calcium uptake (23, 38) and has been shown to reduce mitochondrial calcium levels, neurodegenerative phenotypes and proteostasis defects in sel-12 mutants (16, 23). We found that introduction of an mcu-1 null mutation in the sel-12 mutant background increases puncta formation to levels indistinguishable from wild type animals (Fig. 1G). This suggests that altered mitochondrial calcium signaling is responsible for reducing autophagy in sel-12 mutants. As an alternative method to measure autophagic flux, we immunoblotted for GFP in the GFP::LGG-1 animals to assess lipidation of LGG-1 with phosphatidylethanolamine (PE). When autophagy is induced, PE is conjugated to LGG-1 to anchor it to the autophagosome. Thus, LGG-1-PE detection via western blotting is a reliable method to quantify the rate of autophagy (39). Consistent with this, GFP::LGG-1-PE levels significantly increase in raga-1 null mutants, which have been shown to have elevated autophagy via inhibition of mTORC1 signaling (40) (Fig. S1A,B). Consistent with our autophagosome puncta quantification, we found the amount of processed GFP::LGG-1-PE was significantly decreased in sel-12 mutants, and the level of processed GFP::LGG-1-PE was restored in mcu-1; sel-12 animals (Fig. 1H,I). Therefore, unlike proteasome activity in sel-12 mutants, these data suggest that autophagy is defective, which is consistent with previous observations of disrupted autophagy in sel-12 mutants as well as other models studying presenilin function (6, 9, 16, 41, 42) and also implicate a critical role of mitochondrial calcium in this defect.
Inhibition of mTORC1 in sel-12 mutants increases autophagy
A central inhibitor of autophagy is the serine/threonine protein kinase mTORC1 signaling pathway (43, 44). To investigate whether mTORC1 has a role in inhibiting autophagy in sel-12 mutants, we genetically ablated two key positive mediators of the mTORC1 pathway in sel-12 mutant animals. These include the gene encoding the RagA GTPase ortholog, raga-1, which is critical for the activation of mTORC1; and a gene encoding a key effector protein of mTORC1 signaling, ribosomal protein S6 kinase (rsks-1). Analysis of LGG-1::GFP puncta in raga-1(ok386); sel-12 and rsks-1(ok1255); sel-12 double mutant animals reveals that sel-12 mutants with mTORC1 signaling inhibited, unlike sel-12 mutants alone, show robust accumulation of LGG-1::GFP puncta (Fig. 1C-F). Moreover, consistent with mTORC1 acting as a strong inhibitor of autophagy, blocking mTORC1 signaling resulted in elevated puncta formation and GFP::LGG-1-PE levels in wild type animals (Fig. 1C-F, Fig. S1A,B). Notably, a similar number of puncta is observed in sel-12 mutants with compromised mTORC1 signaling (Fig. 1C-F). These data indicate that sel-12 mutants have the capacity to carry out autophagy when mTORC1 signaling is disrupted. However, without mTORC1 inhibited, autophagy is blunted in sel-12 mutants, suggesting a role of activated mTORC1 in mediating sel-12 phenotypes.
mTORC1 signaling is upregulated in sel-12 mutants.
Given that the mTORC1 pathway is a central inhibitor of autophagy and is a critical metabolic sensor (43, 44) and we have observed increased mitochondrial metabolic activity due to elevated ER to mitochondria calcium signaling in sel-12 mutants (23), we asked whether mTORC1 signaling is elevated in sel-12 mutants. To assess mTORC1 activity, we examined phosphorylation levels of the central mTORC1 target RSKS-1/S6 kinase in sel-12 mutants. Strikingly, phosphorylated RSKS-1 was significantly increased in sel-12 animals compared to wild-type animals (Fig. 2A,B), indicating that mTORC1 signaling is elevated in sel-12 mutants. Next, we sought to determine whether the increased mitochondrial activity observed in sel-12 mutants is leading to the elevation in mTORC1 signaling. Previously, we demonstrated that loss of sel-12 function promotes calcium uptake into the mitochondria from the ER, a process that increases mitochondrial activity and leads to the subsequent proteostatic collapse and neurodegeneration observed in sel-12 mutants (16, 23). Indeed, reducing ER calcium release or mitochondrial calcium uptake in sel-12 mutants, as well as reducing oxidative phosphorylation, suppresses the proteostasis defects and neurodegeneration phenotypes observed in sel-12 mutants (16, 23). To determine whether this altered calcium signaling pathway observed in sel-12 mutants is responsible for mTORC1 hyperactivation, we examined phospho-RSKS-1 levels in mcu-1; sel-12 double mutants and found that introduction of the mcu-1 null mutation leads to reduced phopsho-RSKS-1 levels in sel-12 mutants compared to sel-12 mutants alone (Fig. 2C,D). It is likely that mitochondrial calcium levels are responsible for changes to mTORC1 activation, as the mcu-1 mutation does not increase cytosolic calcium levels relative to the sel-12 mutation alone (Fig. S2). To determine whether mitochondrial hyperactivity due to elevated mitochondrial calcium promotes mTORC1 activation, we treated sel-12 worms with doxycycline to reduce mitochondrial respiration (23, 45). Similar to blocking mitochondrial calcium uptake, sel-12 mutants treated with doxycycline abrogated the increase in phospho-RSKS-1 levels (Fig. 2E,F). Collectively, these data suggest that the altered ER-mitochondrial calcium signaling in sel-12 mutants causes aberrant activation of the mTORC1 pathway by increasing mitochondrial activity.
Reduction of mTORC1 signaling suppresses neuronal defects in sel-12 mutants.
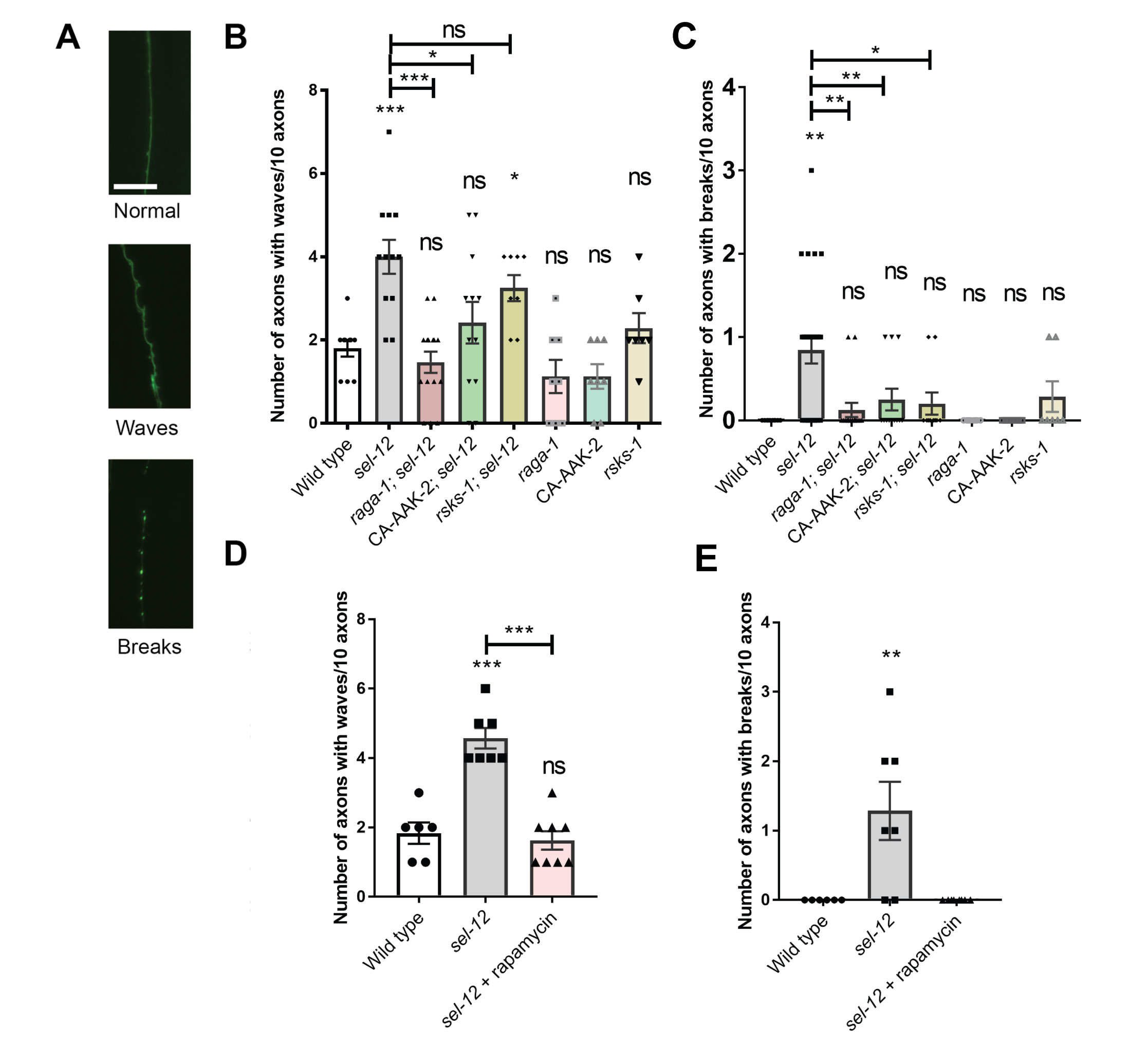
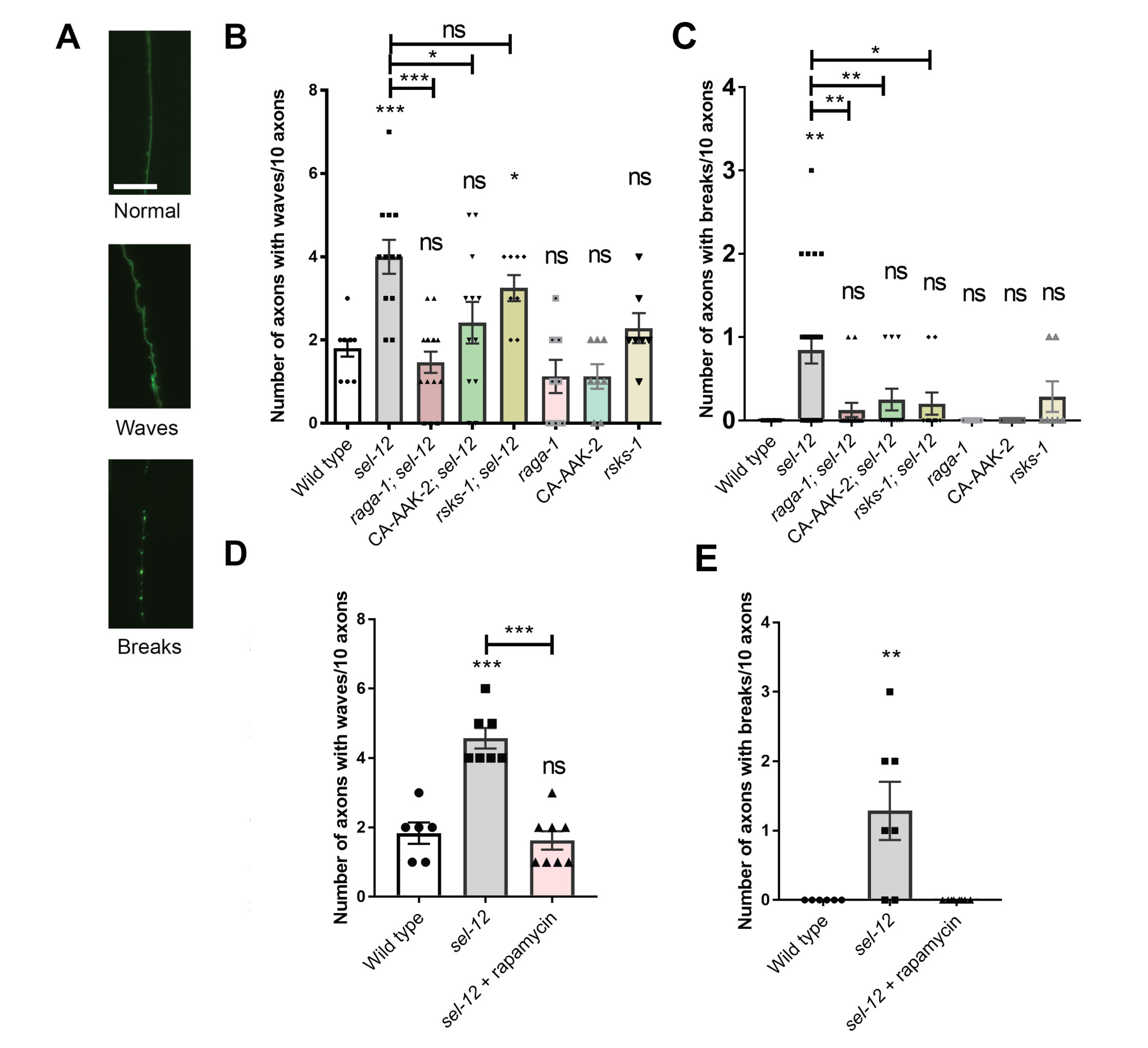
We next asked whether mTORC1 activity contributes to the behavioral and neuronal defects seen with sel-12 loss. We first examined the structure of the C. elegans mechanosensory neurons, which show age dependent structural decline and neurodegeneration (46-48). Previously, we found that these structural aberrations associated with aging develop precociously in sel-12 mutants (23). Day 1 adult sel-12 mutants display numerous ectopic neurite sprouts stemming off the ALM neuronal soma which are absent in wild type animals, and also show defects in the structure of the ALM and PLM axons, exhibiting abnormal lesions at a higher frequency relative to wild-type animals at day 1 (Fig. 3A-C, Fig. S3A,C). To determine whether mTORC1 inhibition can suppress these neuronal morphological defects, we examined mechanosensory neuron structure in rsks-1; sel-12 and raga-1; sel-12 animals, as well as in aak-2(uthIs248); sel-12 animals, which carry a mutation that results in constitutive activation of the catalytic subunit of 5' adenosine monophosphate-activated protein kinase (AMPK/AAK-2), a global energy sensor and a major inhibitor of mTORC1 activity (49). raga-1; sel-12, aak-2(uthIs248); sel-12, and rsks-1; sel-12 animals each showed substantially improved neuronal morphology, with reduced ectopic neurite processes stemming off the soma (Fig. 3B) and reduced frequency of lesions (Fig. 3C), wave-like processes (Fig. S3B) and breaks (Fig. S3C) in the ALM and PLM axons. In addition, we treated the sel-12 animals with rapamycin, a clinical grade drug that specifically inhibits mTORC1, and examined neuronal morphology. Consistent with the genetic manipulations, treatment with rapamycin showed similar improvements to the ALM soma (Fig. 3D) and axonal structure (Fig. 3E, Fig. S3D,E).
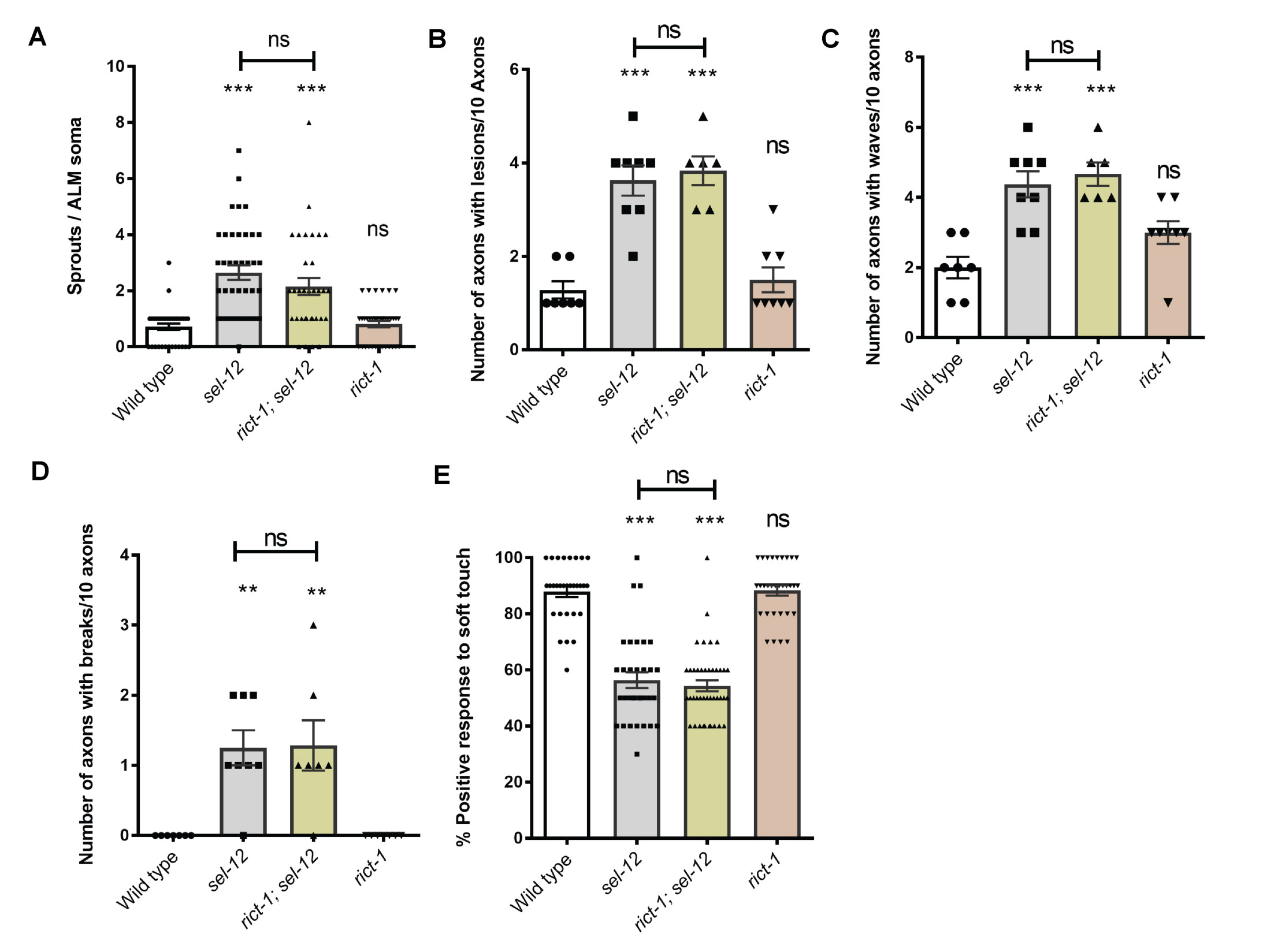
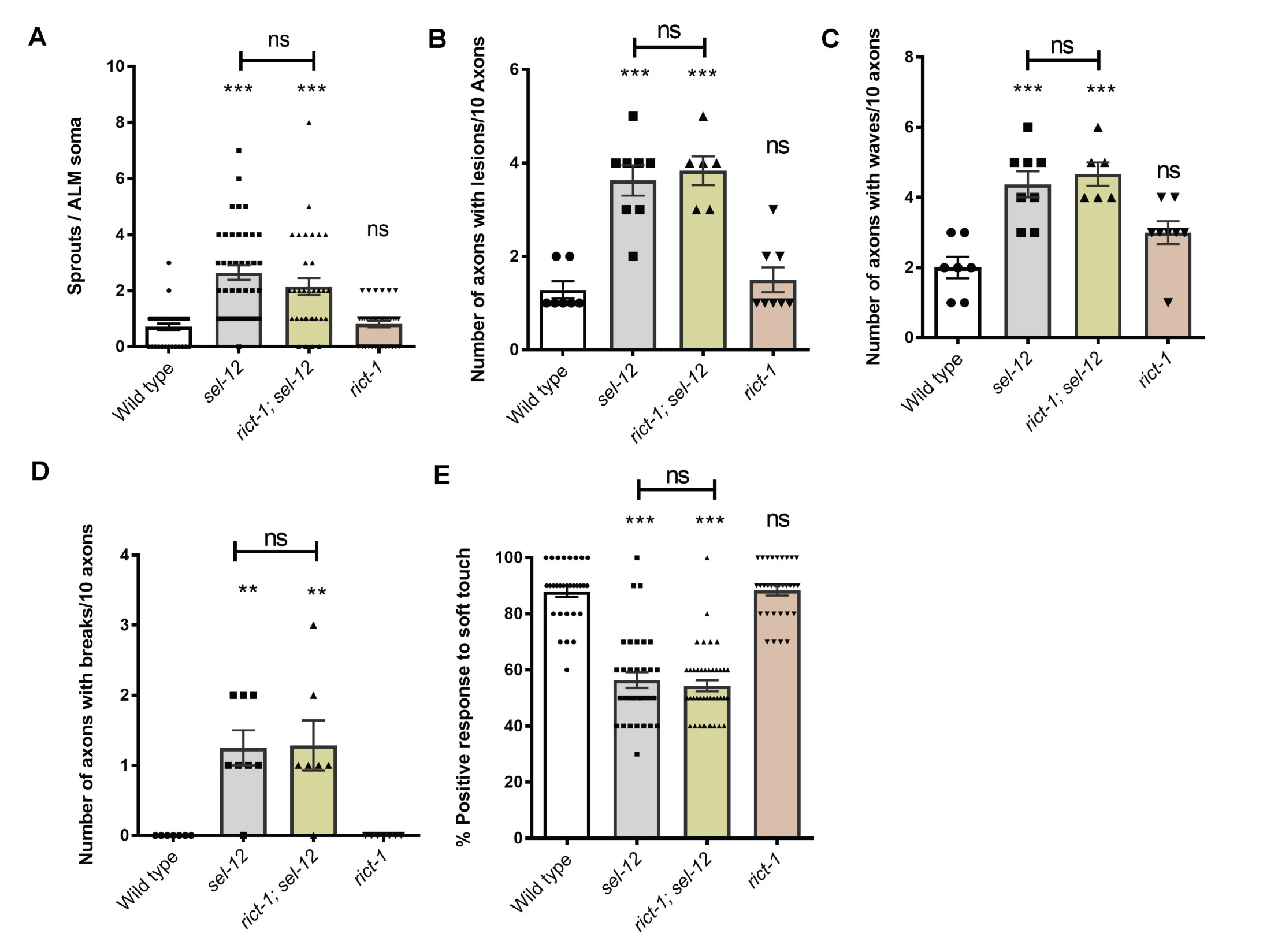
To determine whether the structural improvements we observed in the mechanosensory neurons translates to functional improvement, we examined the effects of genetic and pharmacological mTORC1 inhibition on soft touch behavior, which is controlled by the mechanosensory neurons. Wild type day 1 adult animals, when touched on the anterior or posterior half of the body with an eyebrow hair, will reverse their progression and move away from the stimulus. Consistent with increased morphological defects in the mechanosensory neurons of aged animals, mechanosensation declines with age (47). This reduced response rate happens prematurely in sel-12 mutants and continues to worsen with age (23). Indeed, day 1 adult sel-12 mutants show pronounced defects in soft touch response (Fig. 3F). Consistent with mechanosensory neuronal structural improvements, the raga-1; sel-12, aak-2(uthIs248); sel-12, and rsks-1; sel-12 double mutants all showed significant improvements to soft touch response (Fig. 3F). Moreover, rapamycin treatment recapitulated these improvements (Fig. 3G). Furthermore, this improvement is specific to mTORC1, as inhibition of mTORC2 signaling in sel-12 mutants showed no improvement to sel-12 mutant neuronal defects. Indeed, genetic ablation of RICTOR, rict-1, which is required for the activation of mTORC2, in the sel-12 mutant background did not show significant improvements to mechanosensory neuron morphology or soft touch behavior (Fig. S4). These data indicate that neurodegeneration in sel-12 mutants can be suppressed with mTORC1 inhibition.
Additionally, we found that expression of raga-1 cDNA driven under the pan-neuronal rab-3 promoter (50) was sufficient to fully prevent any improvements to soft touch behavior in raga-1; sel-12 animals (Fig. 3H), indicating a cell autonomous role of mTORC1 hyperactivity in the nervous system of sel-12 mutants. Additionally, unlike in wild type animals, pan-neuronal expression of raga-1 in the neurons of sel-12 animals significantly aggravated their touch defect. These data demonstrate a central role for neuronal mTORC1 activity in mediating and exacerbating the neurodegenerative behavioral defect in sel-12 mutants.
Inhibition of mTORC1 improves proteostasis in sel-12 mutants.
mTORC1 activity may further explain impaired proteostasis in sel-12 animals. In fact, many studies show that modulation of mTORC1 activity widely impacts proteostasis (51). To define a role of mTORC1 in the collapse of proteostasis in sel-12 mutants (16), we first examined animals with body wall expression of polyglutamine (polyQ) Q35::YFP fusion protein (rmIs132), which aggregates as the animals age (52). While expression of Q35::YFP remains soluble and evenly distributed in day 3 adult wild type animals, adult sel-12 mutants show premature Q35 aggregation by day 3. However, analyses of raga-1; sel-12 and rsks-1; sel-12 animals show a significant reduction in polyQ aggregates at day 3 compared to sel-12 mutants, suggesting that mTORC1 inhibition improves proteostasis in these animals (Fig. 4A,B). Consistent with these results, rapamycin treatment also reduced polyQ aggregates in day 3 adult sel-12 adult animals (Fig. 4C).
As an alternate method to assess the state of proteostasis in sel-12 mutants, we examined animals expressing human Aβ1-42 (dvIs100), which generates proteostatic stress and causes progressive paralysis in the transgenic animals (15). Previously, we found that that sel-12 mutants expressing Aβ1-42 have severely reduced motility relative to either mutant background alone, suggesting that the sel-12 mutation promotes Abeta1-42 toxicity and enhances proteostasis defects (16). To determine the effect of mTORC1 inhibition on motility in this background, we examined swimming behavior of raga-1; sel-12 and rsks-1; sel-12 mutants expressing Aβ1-42, and found they had significantly higher motility compared to sel-12 mutants expressing Aβ1-42 (Fig. 4D). Furthermore, treating sel-12 mutants expressing Aβ1-42 with rapamycin showed similar improvements in motility (Fig. 4E).
To evaluate the state of proteostasis of endogenous proteins, we subjected animals to heat stress (exposure to 37°C) to induce protein misfolding (53) and then examined animal survival. Previously, we found that sel-12 mutants have reduced resistance to heat stress and reduced survival (16). Thus, we examined the survival rate of wild type, sel-12 and raga-1; sel-12 mutants, as well as rapamycin treated sel-12 animals after 1, 2, 3, and 4 hours of exposure to 37°C. We found that the survival rate after heat stress at each time point was increased in the sel-12 mutants with mTORC1 signaling inhibited either genetically or pharmacologically relative to sel-12 mutants (Fig. 4F,G). Altogether, these data suggest that mTORC1 impacts proteostasis in sel-12 mutants and that the defects in proteostasis due to loss of SEL-12 are improved through mTORC1 inhibition.
Improvements to proteostasis and neuronal function through mTORC1 inhibition require the induction of autophagy.
To further investigate the mechanism by which mTORC1 contributes to proteostasis and neuronal defects in sel-12 mutants, we first examined the condition of mitochondrial morphology in sel-12 mutants with mTORC1 signaling abrogated. Previously, we found that sel-12 mutants have severe defects in mitochondrial morphology and function due to elevated mitochondrial calcium (23, 54). To determine whether mTORC1 activity contributes to mitochondrial disorganization in sel-12 mutants, we examined sel-12 and raga-1; sel-12 mutants expressing a mitochondrial localization signal tagged with GFP in the mechanosensory neurons (23, 55, 56). We found that compared to sel-12 mutants, raga-1; sel-12 double mutants had significantly improved mitochondrial organization, suggesting that elevated mTORC1 activity contributes to aberrant mitochondrial structure in sel-12 mutants (Fig. 5A,B).
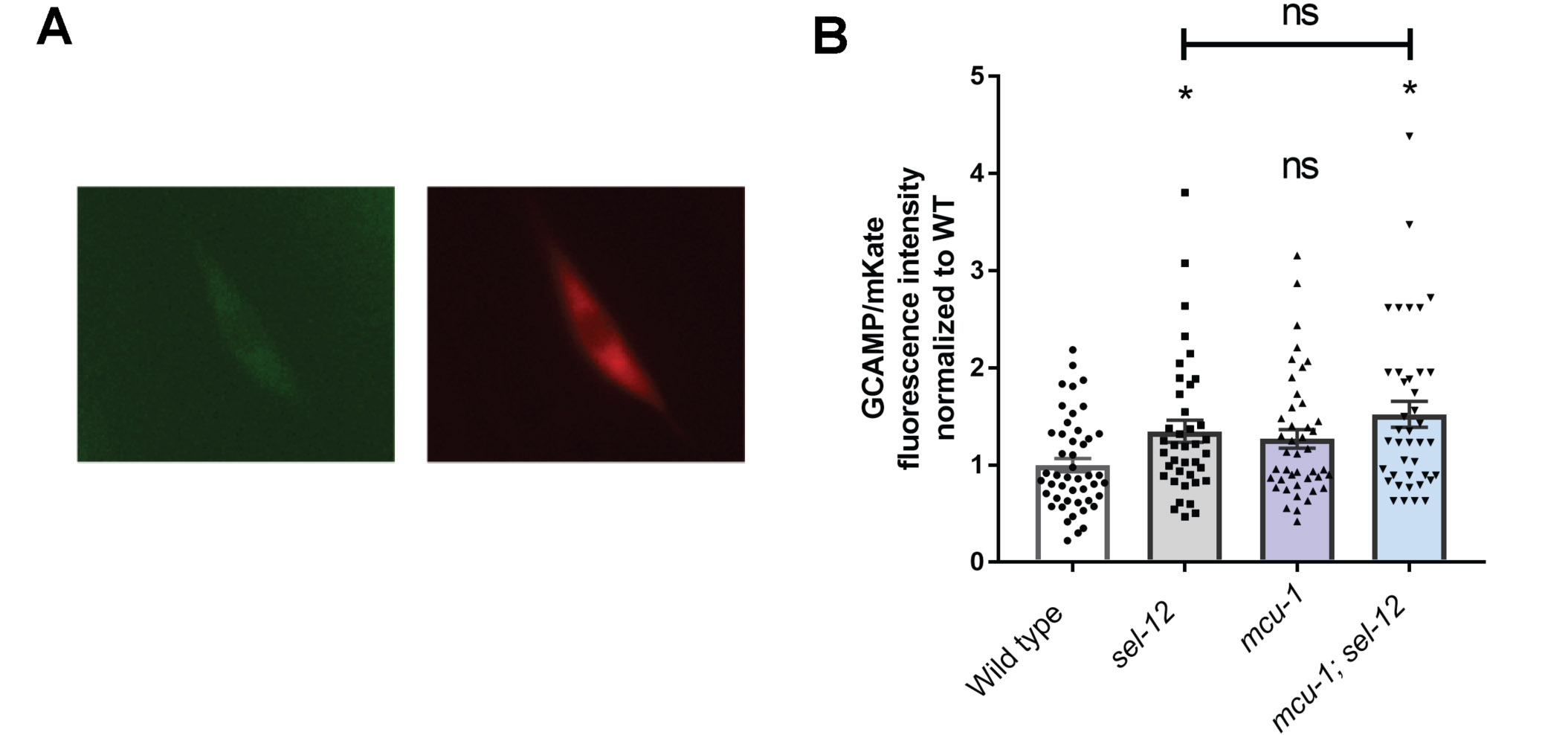
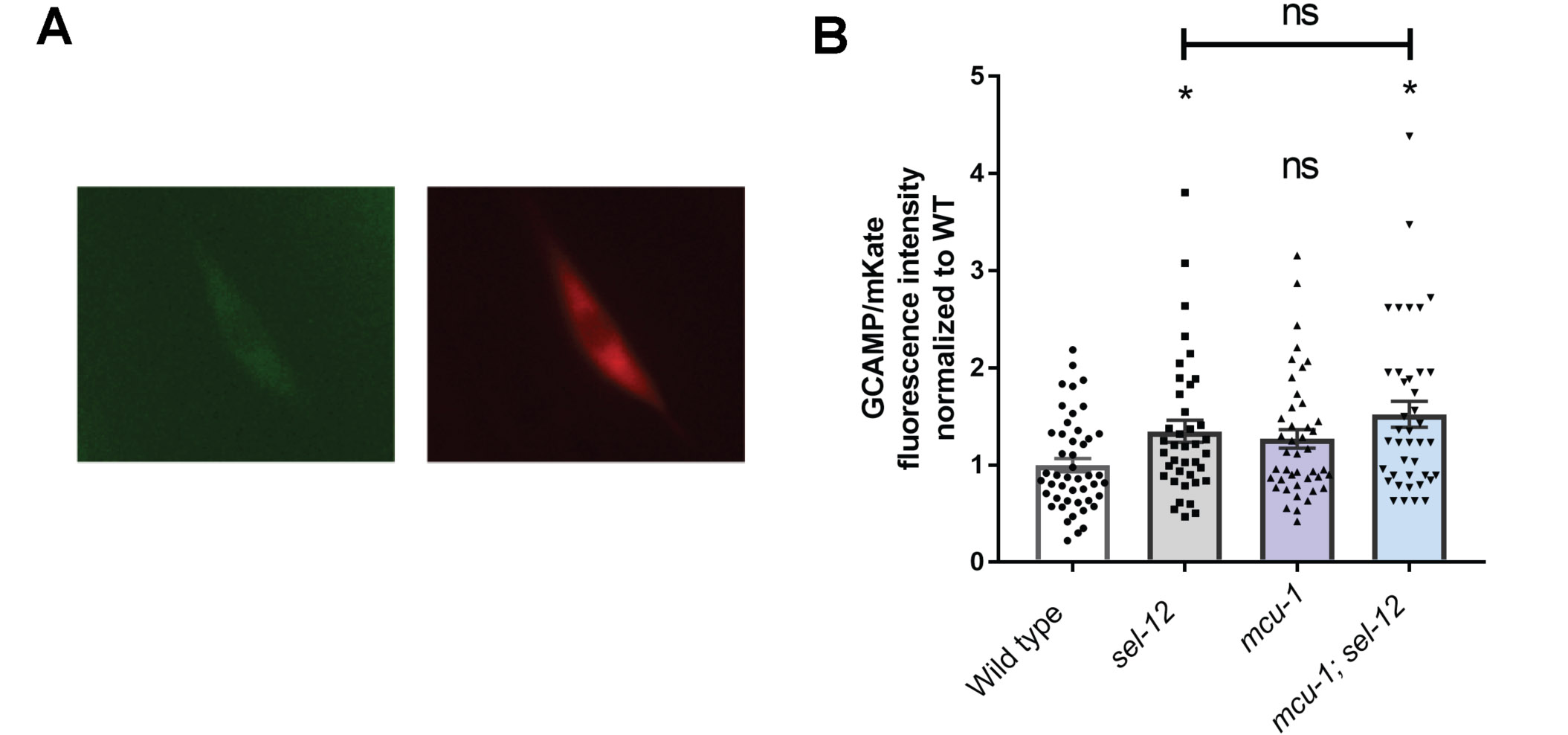
Next, since we previously found that disorganized mitochondrial structure in sel-12 mutants is caused by elevated ER to mitochondrial calcium signaling (23, 54), we asked whether the improvements we observed in raga-1; sel-12 mutants are due to reduced ER-mitochondrial calcium signaling. Using a genetically encoded GCaMP6 calcium indicator localized to the mitochondrial matrix (23), we assessed mitochondrial calcium levels in the mechanosensory neurons. Notably, we found that calcium levels were unchanged after mTORC1 inhibition (Fig. 5C,D), indicating that mTORC1 activation is likely a downstream consequence of the altered calcium signaling observed in sel-12 mutants. This data is consistent with our pRSKS-1 western blot data showing that reduction of mitochondrial calcium uptake reduced pRSKS-1 levels in the sel-12 mutant background (Fig. 2C,D).
Thus, since mTORC1 is likely not playing a role in mitochondrial calcium signaling in the sel-12 mutants, it is possible mTORC1’s inhibition of autophagy is responsible for the defects in proteostasis and neuronal function observed in sel-12 mutants. To determine whether the improvements in behavior and proteostasis through mTORC1 inhibition is primarily due to promoting autophagy, we knocked down inducers of autophagy lgg-1 and bec-1 using RNA interference (RNAi). Importantly, RNAi directed to lgg-1 or bec-1, which encodes the C. elegans beclin 1 ortholog, have been shown to inhibit autophagy (37, 57). From these analyses, we found that the improvements to soft touch behavior in raga-1; sel-12 mutants is abrogated when treated with lgg-1 or bec-1 RNAi (Fig. 6A), and these animals resemble sel-12 mutant animals. Similarly, the improvements to proteostasis as measured by the number of Q35 aggregates in raga-1; sel-12 double mutants are lost with either lgg-1 or bec-1 RNAi treatment (Fig. 6B). Additionally, the increase in swimming rate is lost in raga-1; sel-12 animals expressing Aβ1-42 when treated with lgg-1 or bec-1 RNAi (Fig. 6C). These data suggest mTORC1 primarily impacts proteostasis and neuronal function in sel-12 animals by inhibiting autophagy. Altogether, our data identify activation of mTORC1 as a critical pathway by which SEL-12 loss results in neurodegeneration, and define an important role of the mTORC1 pathway in exacerbating the defects in proteostasis and autophagy following loss of SEL-12.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}