Family and clinical examination
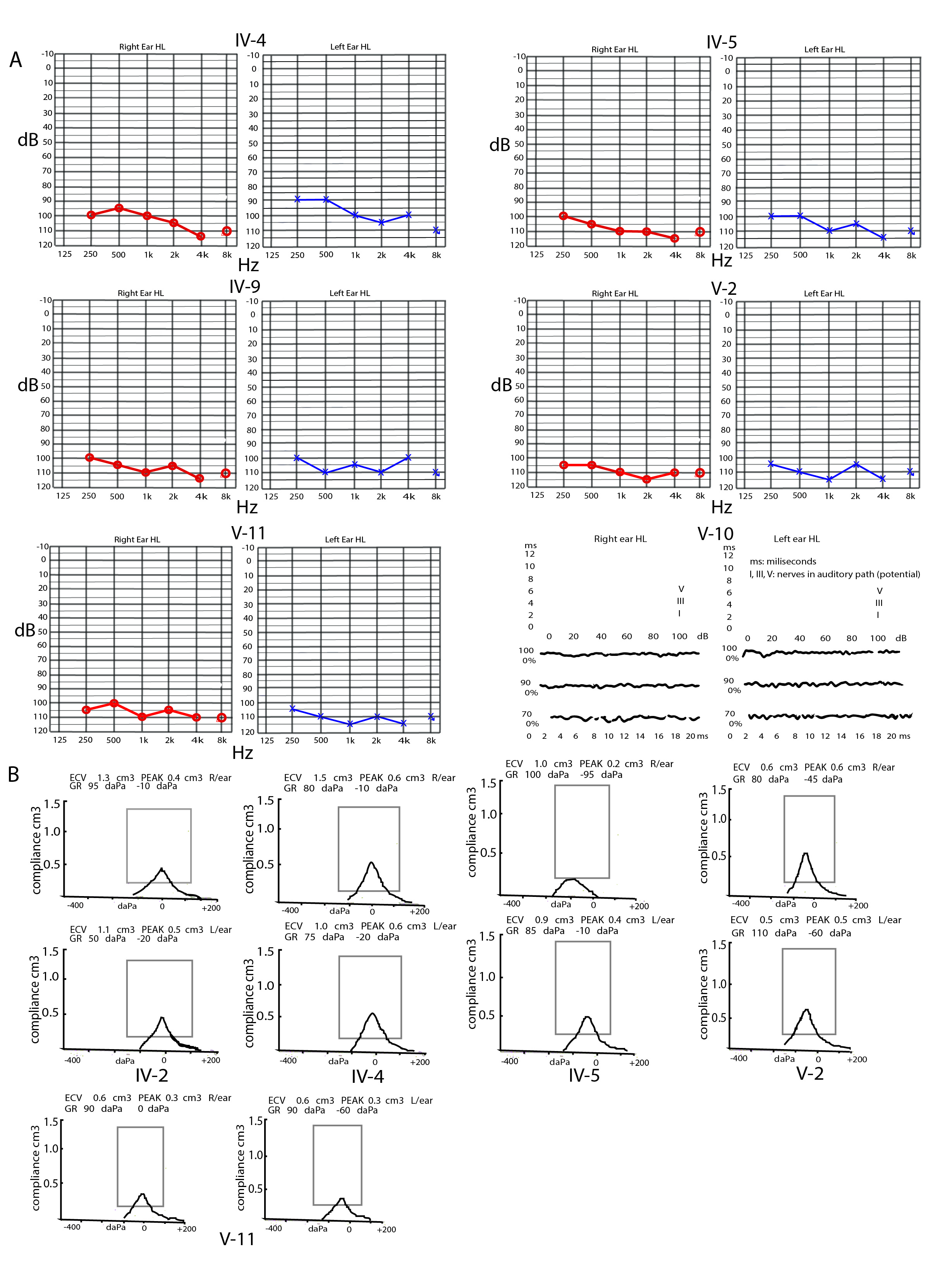
The consanguineous family, recruited from rural suburbs in southern Pakistan, included the two phenotypes of intellectual disability and deafness, but none of the patients showed both phenotypes. Subjects with intellectual disability showed axonal polyneuropathy and other physical features[16]. There were seven deaf patients (four males and three females) in this five-generation pedigree. The affected subjects segregated in the fourth and fifth generations in a manner that suggested an autosomal recessive inheritance pattern (Fig. 1A). The clinical evaluations included pure-tone audiometry (PTA) at 125-8 kHz in a quiet room and impedance audiometry or tympanometry of six affected individuals (IV-2, IV-4, IV-5, IV-9, V-2, V-11) and one unaffected individual (IV-16). A brain stem evoked response audiometry (BERA) test was performed for subject V-10 (Additional File 1). Written informed consent was obtained from the family elders under the regulations of the Institutional Review Board of the School of Life Sciences, Central South University, and Quaid-i-Azam University Islamabad.
Whole-exome sequencing (WES) and data analysis
Blood samples were collected from 7 affected and 28 unaffected subjects in the family in 5 ml EDTA tubes (Fig. 1A). Genomic DNA was extracted from leukocytes of all recruited family members using the Wizard Genomic DNA Purification Kit (Promega A1620). The quality of DNA was checked on a Nanospec Cube Biophotometer (Nanolytik, Dusseldorf, Germany). Whole-exome sequencing (WES) was performed on one microgram genomic DNA samples from two affected (V-2, V-11) and one unaffected individual (IV-16) using the Agilent Sure-Select Target Enrichment System (V6) and the Illumina X Ten platform. Generated reads were aligned to human reference genome GRCH37/hg19 using Burrows-Wheeler Aligner (version 0.7.12-r1044). Variants were called by GATK (version 3.5) and annotated by ANNOVAR (version 2015-06-17). We filtered WES data variants as the strategy in Ahmed et al[16] and excluded the variants with allele frequencies > 0.01 as reported in the GnomAD, Exome Aggregation Consortium (ExAC), 1000 Genome and ESP6500 databases. Missense mutations, nonsense mutations and splicing variants were evaluated. Sorting Intolerant from Tolerant (SIFT), Polymorphism Phenotyping V.2 (PolyPhen-2), and Mutation Taster were used to assess the overall impact of the variant selected for Sanger analysis. We have uploaded the associated datasets of this study to the SRA - NCBI repository (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA675842&o=acc_s%3Aa).
Sanger sequence analysis of the selected gene variant
Primers were designed with Primer3 to validate the selected variant (NM_194248: c.3289-1G > T) from WES data through Sanger sequencing. The primer sequences were as follows: forward, 5′-cgtggtccttagggggttt-3′; reverse, 5′-gagtgcgacttggtgcagat-3′. PCR amplification was performed with these primers for all available individuals by using the GeneAmp PCR system 2720 (Applied Biosystems, Foster City, CA, USA). The PCR mix consisted of 5 µL of premix ExTaq Polymerase (Takara Bio, Dalian, China), 30 ng of DNA, 50 ng of primers, and ddH2O for a total volume of 10 µL. The PCR conditions were as follows: an initial denaturing step at 95℃; 35 cycles of denaturing at 95℃ for 1 min, annealing at 58℃ for 1 min, and extension at 72℃ for 1 min; and a final extension step at 72℃ for 10 min. The PCR products were verified by 1% polyacrylamide gel electrophoresis and ethidium bromide (EB) staining, after which Sanger sequencing was performed. Sequence traces were visualized with CodonCode Aligner (Version 7.1.2) and SeqMan (Lasergene).
Minigene design and plasmid construction
A minigene splicing assay was performed as previously described[17]. In order to assess the impact of the detected sequence variant on splicing, a minigene assay was performed for exons 25–29 of OTOF (Fig. 3A). The primers were designed with Primer-Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast), and the sequences were as follows: forward, 5′-TTGGCGCGCCCTCTGGCTTTGAGAGCACCAG-3′; reverse, 5′-ATAAGAATGCGGCCGCAGCCTGACTGGACAGATGGAT-3′. The genomic DNA (gDNA) of the unaffected heterozygous individual IV-16 was amplified using Phanta Max Super-Fidelity DNA polymerase (p505-d1/d2/d3, Vazyme, Nanjing, Jiangsu, China) with the appropriate primers and then introduced into the pCAGGS-IRES-green fluorescent protein (GFP) vector by Sgsl and NOT1 (ER1892 and FD0596, Thermo Fisher Scientific, Waltham, MA, USA). The plasmid was used to transform E. coli DH5α (CB101-2, Tiangen, Beijing, China). Ten positive clones were selected on Luria-Bertani (LB) plates, and the genotypes of the clones were confirmed by Sanger sequencing. After sequence alignment, wild-type and mutant plasmids were selected to transfect cell lines.
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells and HeLa cells were maintained in DMEM (Gibco-BRL Life Technologies, Grand Island, NY) containing 10% fetal bovine serum (FBS) (Gibco-BRL Life Technologies). Cells were grown in 24-well plates at 37 °C in a 5% CO2 atmosphere. Wild-type and mutant plasmids in an otherwise similar construct were transfected into HEK 293 cells and HeLa cell using Lipofectamine 3000 reagent (L3000008, Invitrogen, Carlsbad, CA, USA).
Reverse transcription PCR
After 24 hours, RNA was extracted from the cells using the FastPure Cell/Tissue Total RNA Isolation Kit (RC101-01, Vazyme, Nanjing, Jiangsu, China). cDNA was generated from 500 ng of total RNA using a RevertAid First Strand cDNA Synthesis Kit (K1621, Thermo Fisher Scientific, Waltham, MA, USA). The primer sequences were as follows: forward, 5′-GTGCTGAATGAGACCCTGTG-3′; reverse, 5′-CCGTGGTGTTCCAGCTGGGG-3′. PCRs were generated using Phanta Max Super-Fidelity DNA polymerase (p505-d1/d2/d3, Vazyme). The amplification was performed for 35 cycles of 72 °C for 1 min, 62 °C for 30 sec, and 72 °C for 1 min.
Construction of the pMD-19T plasmid
The final product was introduced into the plasmid vector pMD19-T using a DNA ligation kit (D102A, Thermo Fisher Scientific, Waltham, MA, USA). Then, the plasmid was used to transform DH5α cells (CB101-2, Tiangen, Beijing, China). The positive clones were selected by blue/white screening on Luria-Bertani (LB) plates; ten successful clones from HEK 293 cells and ten from HeLa cells were confirmed by Sanger sequencing.
{kind=link}