Flowchart of mutational signatures stabilization and clinical characteristics.
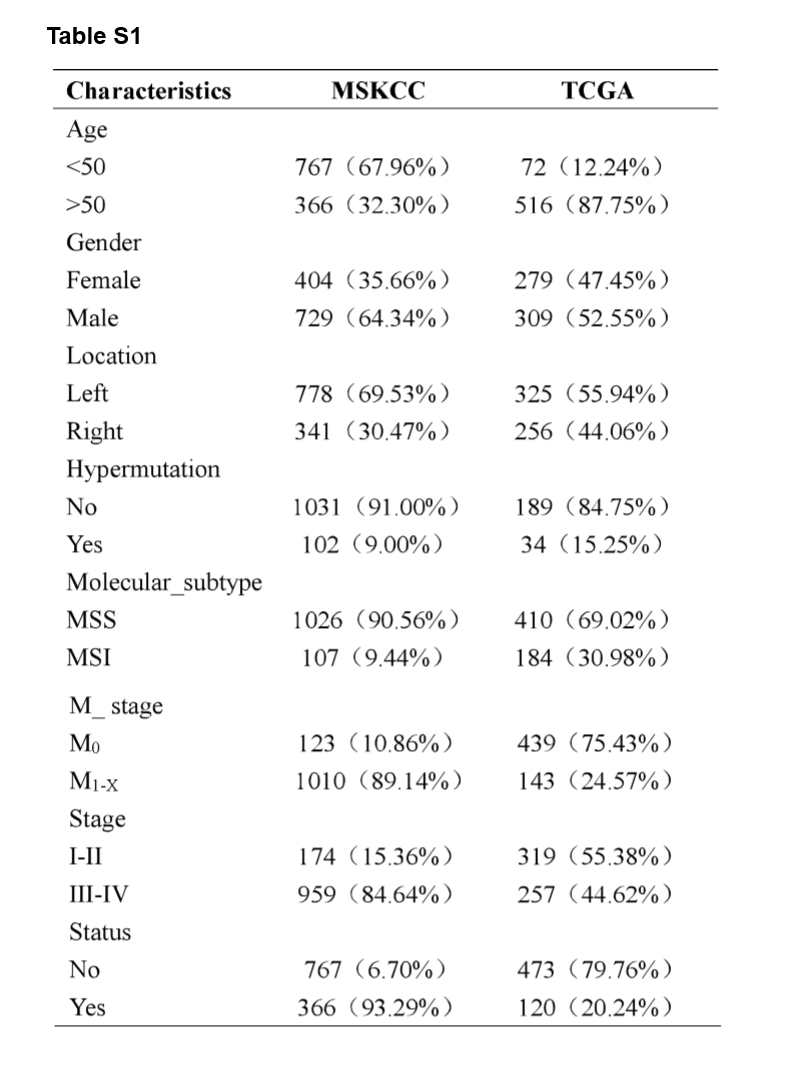
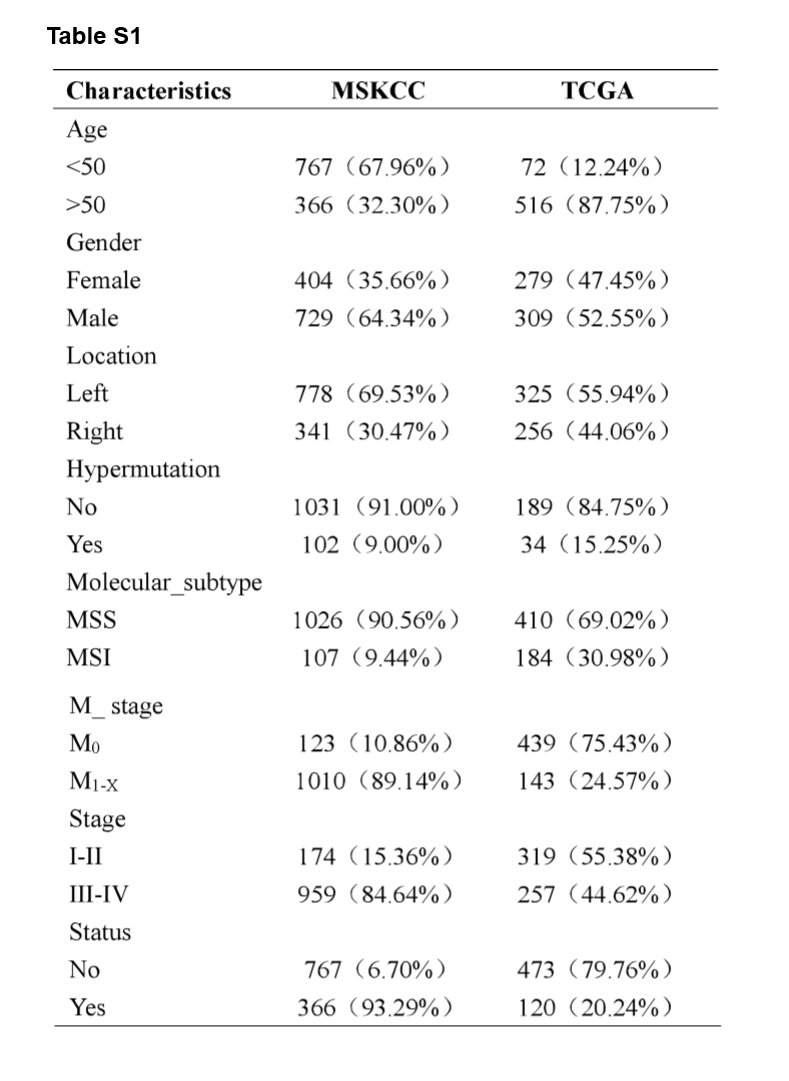
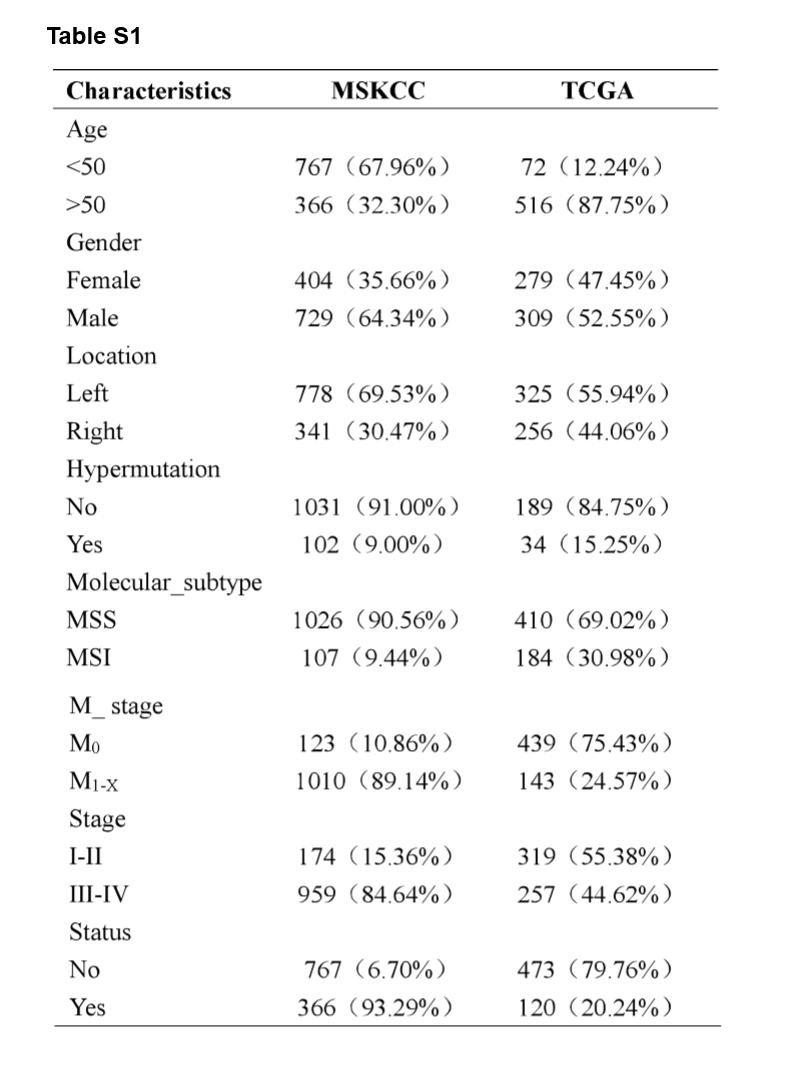
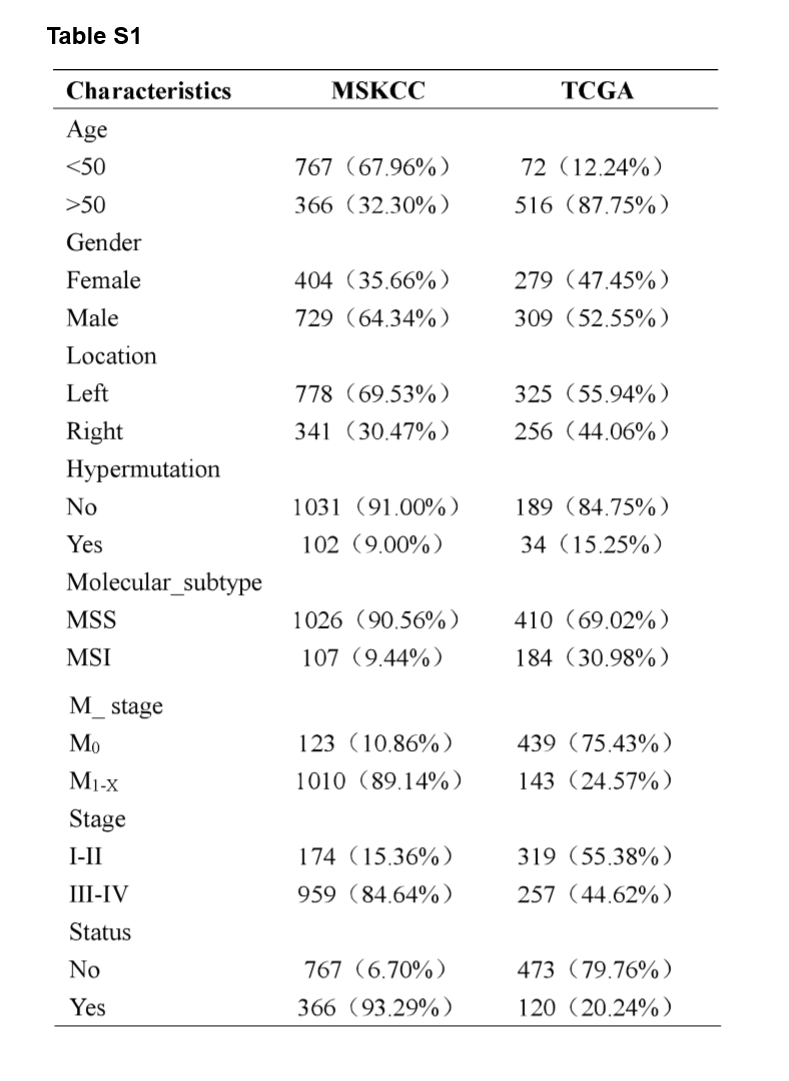
A flowchart demonstrated the procedure how to analyze the mutational signatures and their correlation with clinical characteristics and immunity, as well as prognostic models based on the mutated genes is shown in Fig. 1. This study obtained data from two cohort studies, Memorial Sloan Kettering Cancer Center (MSKCC) database (n = 1,133) and The Cancer Genome Atlas (TCGA) database (n = 588), respectively. The training cohort, MSKCC cohorts, comprised 729 male patients (64.34%) and 404 female patients (35.66%), while the validation cohort, TCGA cohort, comprised 309 male patients (52.55%) and 279 female patients (47.45%). The clinical characteristics of all CRC patients were listed in Supplementary Table S1.
Construction and verification of the mutational signature prognostic classifier.
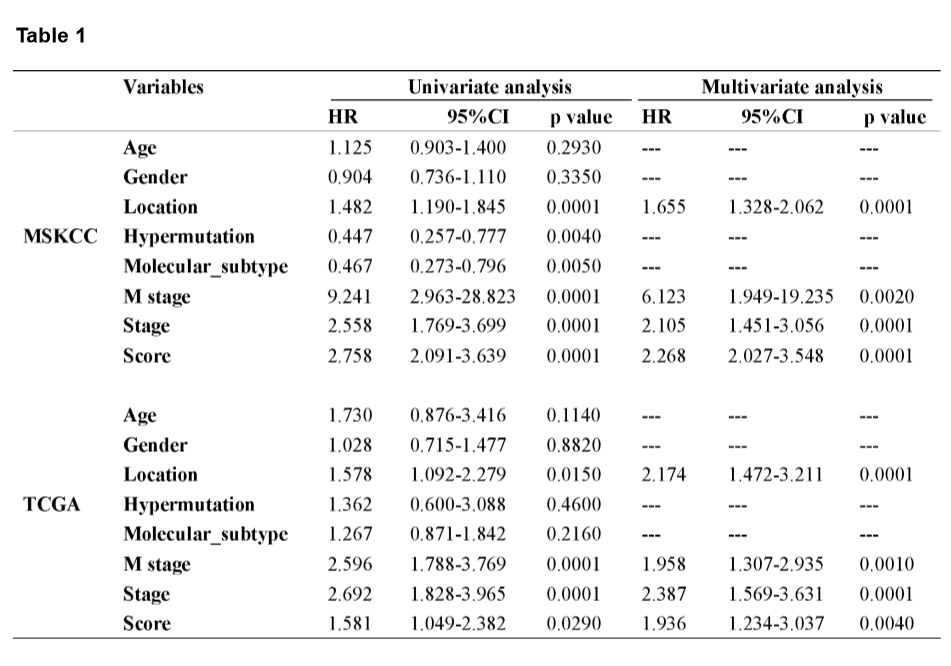
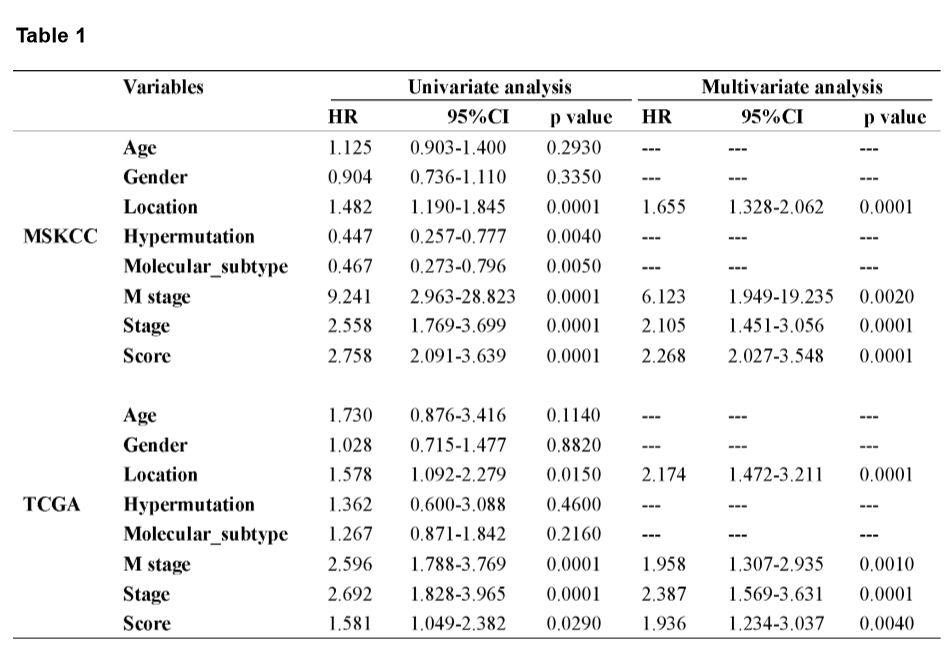
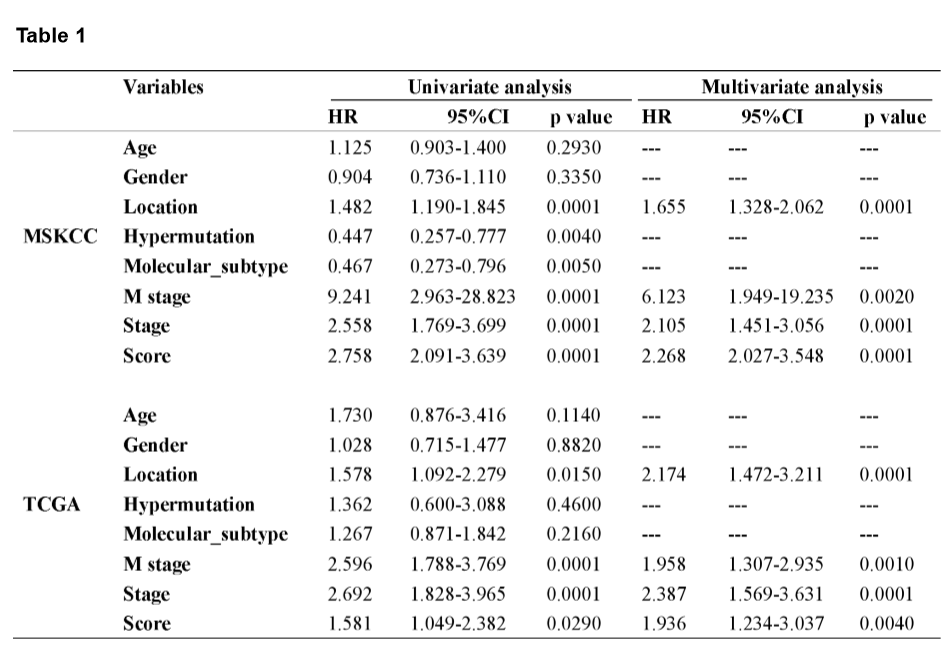
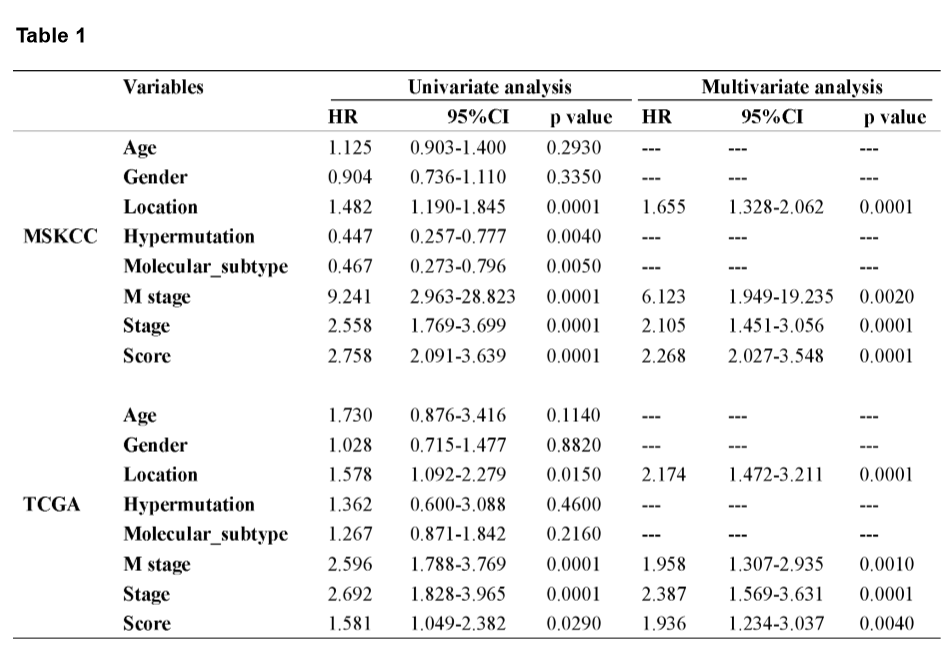
A mutational signature including 521 genes was identified to be related to survival in CRC by MSKCC cohort, and 27 mutated genes which were included in the classifier were identified by least absolute shrinkage and selection operator (LASSO) analysis (Fig. 2A and B). The coefficients of the 27 mutated genes were shown in Table S2. Using Kaplan-Meier analysis, the mutational signature effectively stratified CRCs into high- and low-risk groups, and the classified high-risk group showed a poorer OS compared with the low-risk group, which verified in both MSKCC and TCGA cohorts (Fig. 2E and F). The receiver operating characteristic (ROC) curves indicated that the classifier had a strong predictive ability, as the area under the curve (AUC) values for 1-, 3-, and 5-year OS were 0.712, 0.670, and 0.682, respectively (Fig. 2G). To assess the predictive power of the classifier, we compared the area under ROC curves between the classifier and risk score, tumor location, M stage, and TNM stage. The result showed that the classifier had a better predictive power and accuracy than other clinical features (Fig. 2H). In multivariate Cox analysis of both the MSKCC and TCGA cohorts, the classifier was identified as an unfavorable prognostic factor (Fig. 2I and J). The results in the univariate and multivariate analyses of prognostic factors shown in Table 1 revealed mutational signature is an independent, unfavorable prognostic indicator for CRC in both the MSKCC and TCGA cohorts.
Construction of a predictive nomogram in CRC.
Using the data of the training cohort, a nomogram was generated to predict the OS (Fig. 3A). The predictors included tumor location, M stage, TNM stage, and risk score, among which the risk score had the highest C-index. The calibration plots for the 3- and 5-year OS were well predicted in the training cohort (C-index = 0.666) and the validation cohort (C-index = 0.689) (Fig. 3B and C). The predictive power of the nomogram comprising the mutational signature was compared to clinicopathological risk factors using ROC analysis. The result indicated that the OS was more accurately predicted by the nomogram than by the risk factors in both cohorts (Fig. 3D).
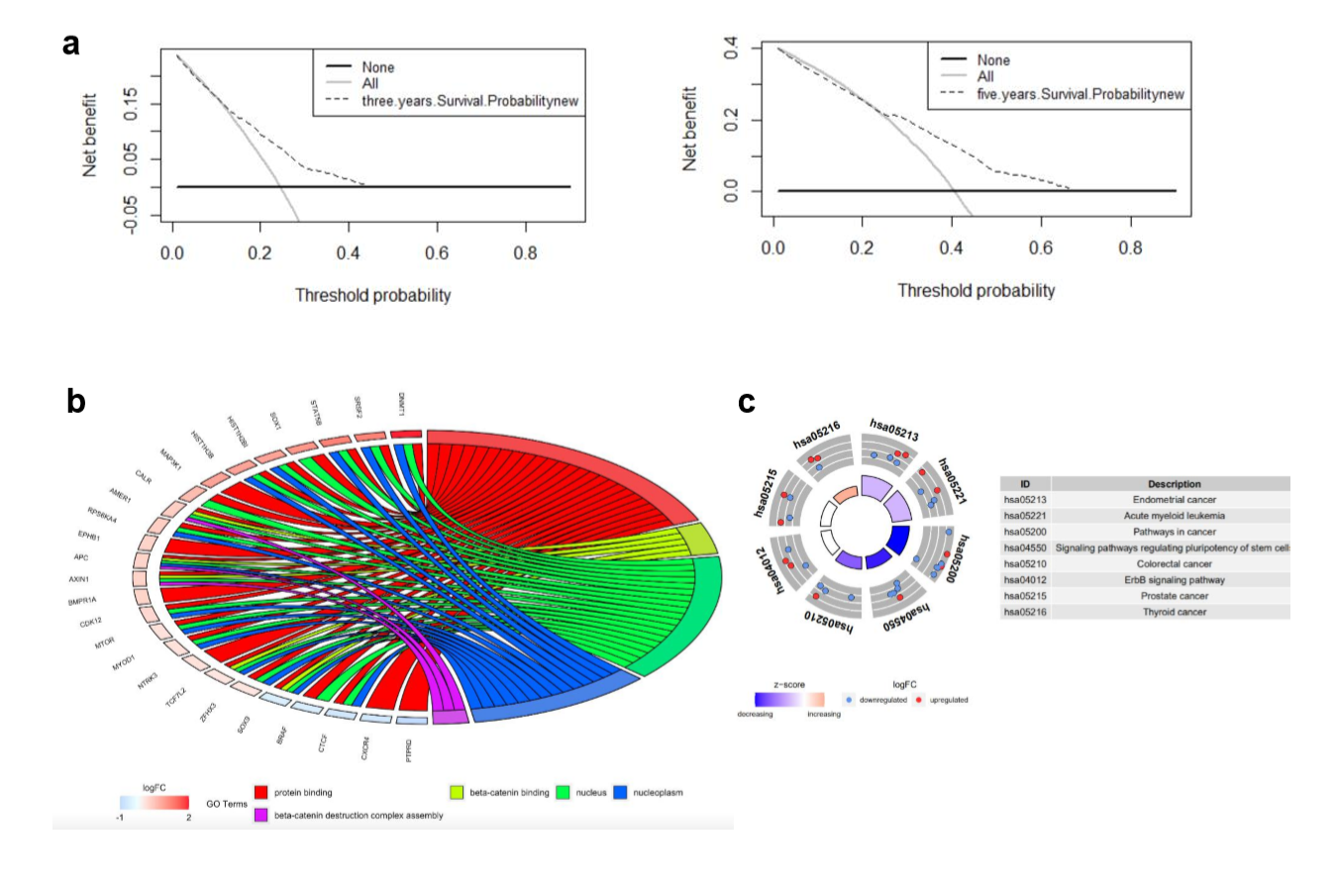
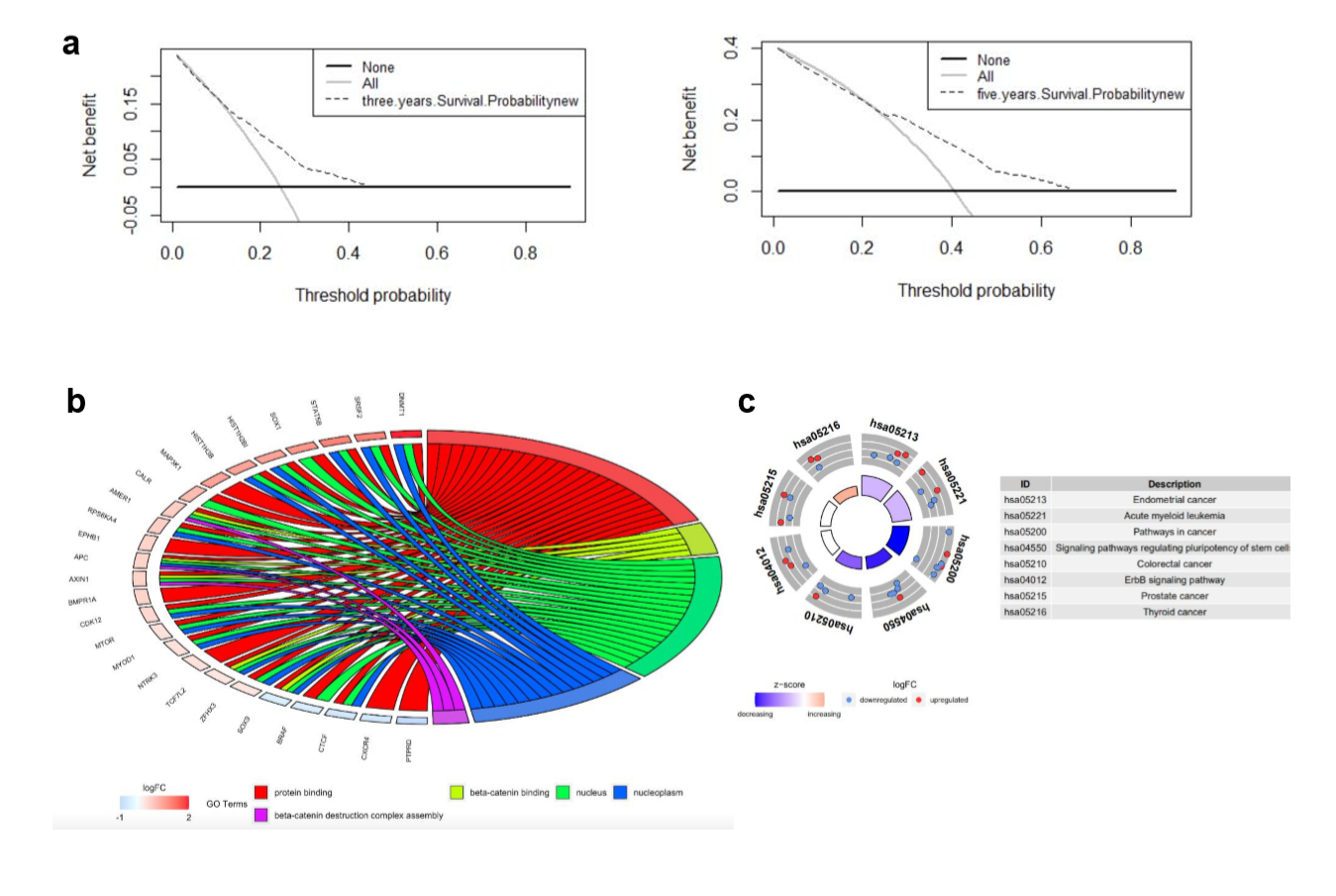
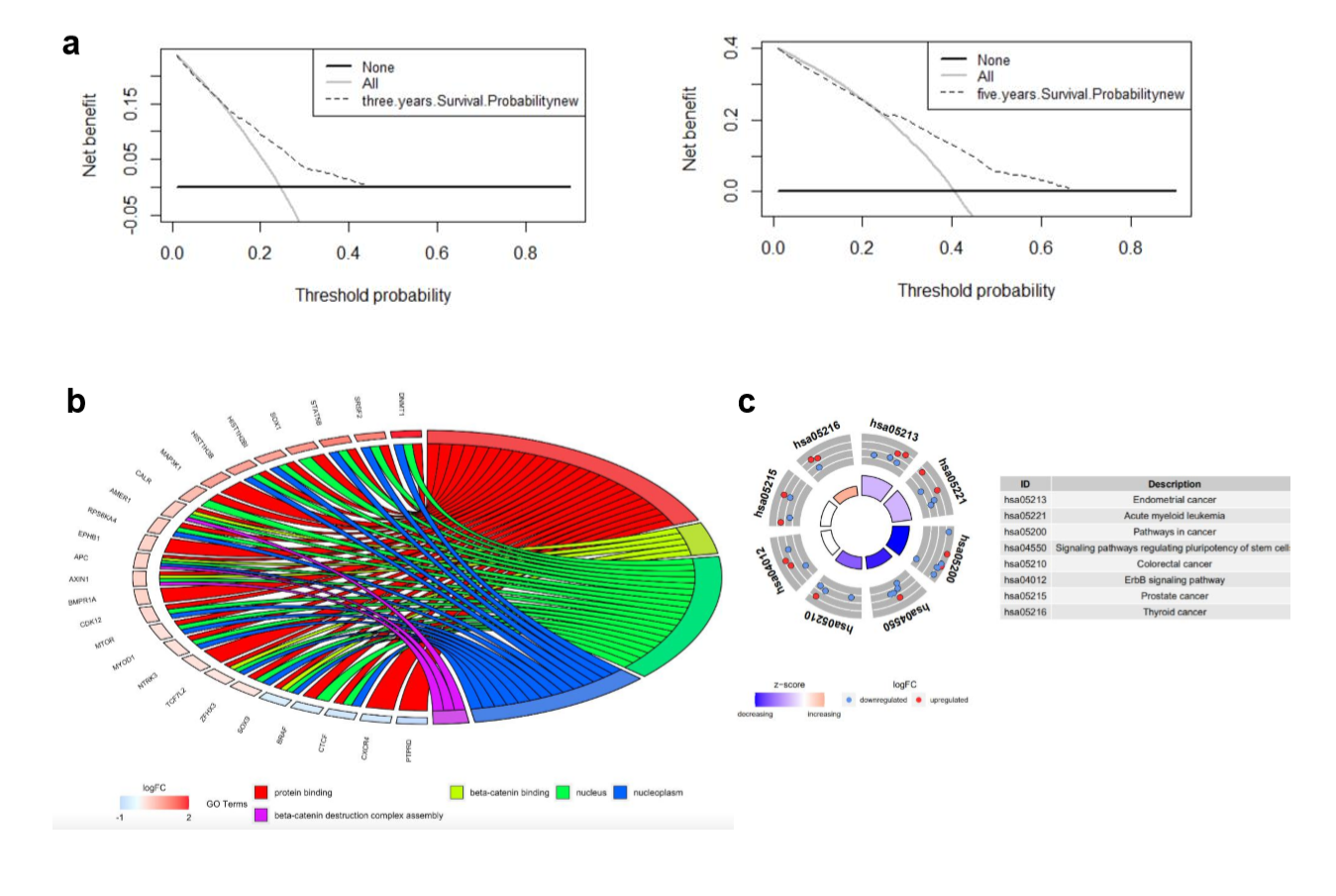
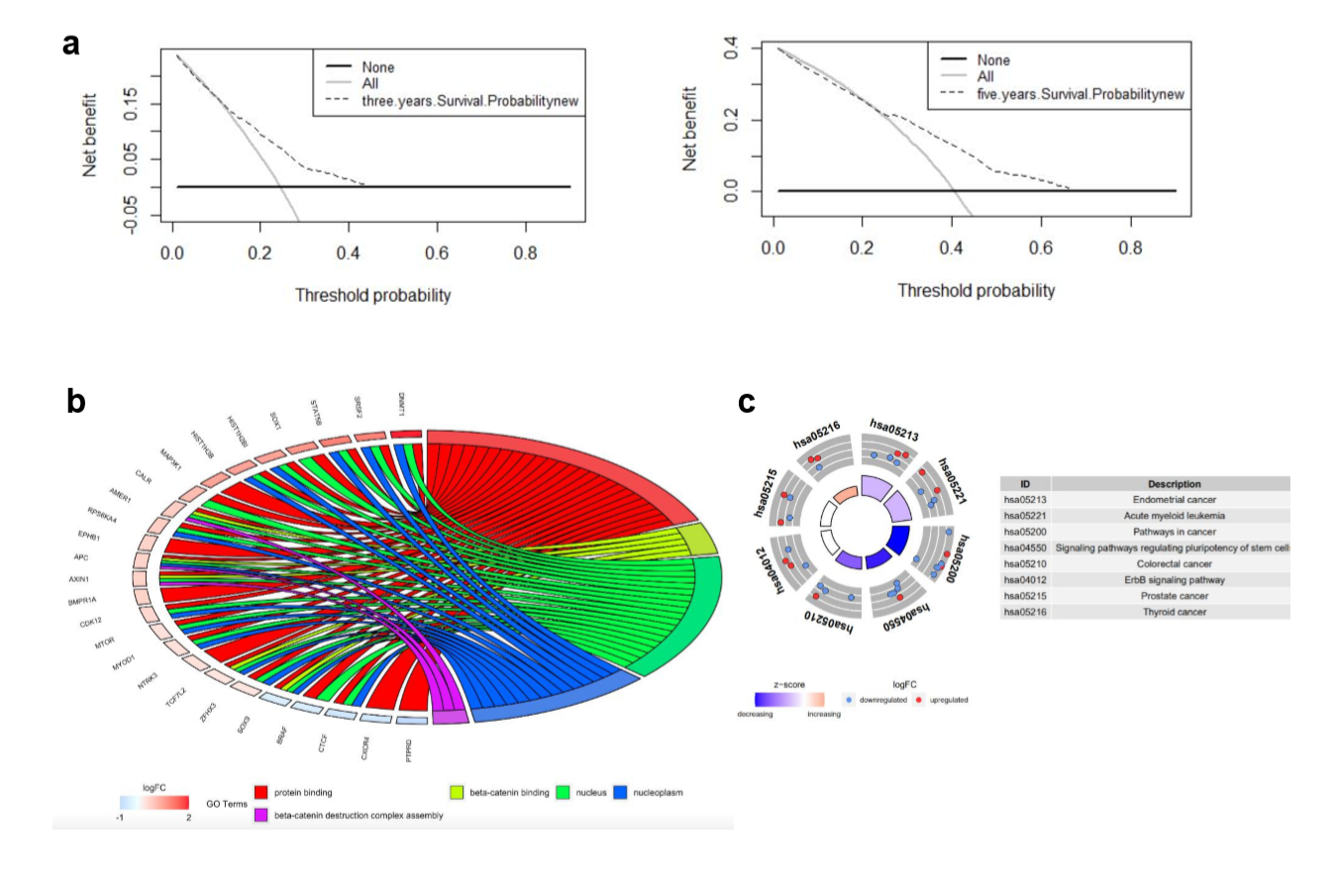
Decision curve analysis was used to quantify clinical application by net benefits at different threshold probability in our nomogram model. Here we found that threshold probabilities of 0 ~ 0.43 and 0 ~ 0.65 were the most beneficial for predicting 3- and 5-year OS, respectively (Supplementary Fig. S1A). Gene ontology (GO) analysis based on the 27 mutated genes demonstrated that mutated genes were mainly enriched in protein binding, beta-catenin binding, nucleus, nucleoplasm, and beta-catenin destruction complex assembly (Supplementary Fig. S1b). Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses revealed that these 27 mutated genes were associated with endometrial cancer, acute myeloid leukemia, pathways in cancer, signaling pathways regulating pluripotency of stem cells, CRC, ErbB signaling pathway, prostate cancer, and thyroid cancer (Supplementary Fig. S1C).
Mutational landscape of significantly mutated genes in defined high- and low-risk subgroups.
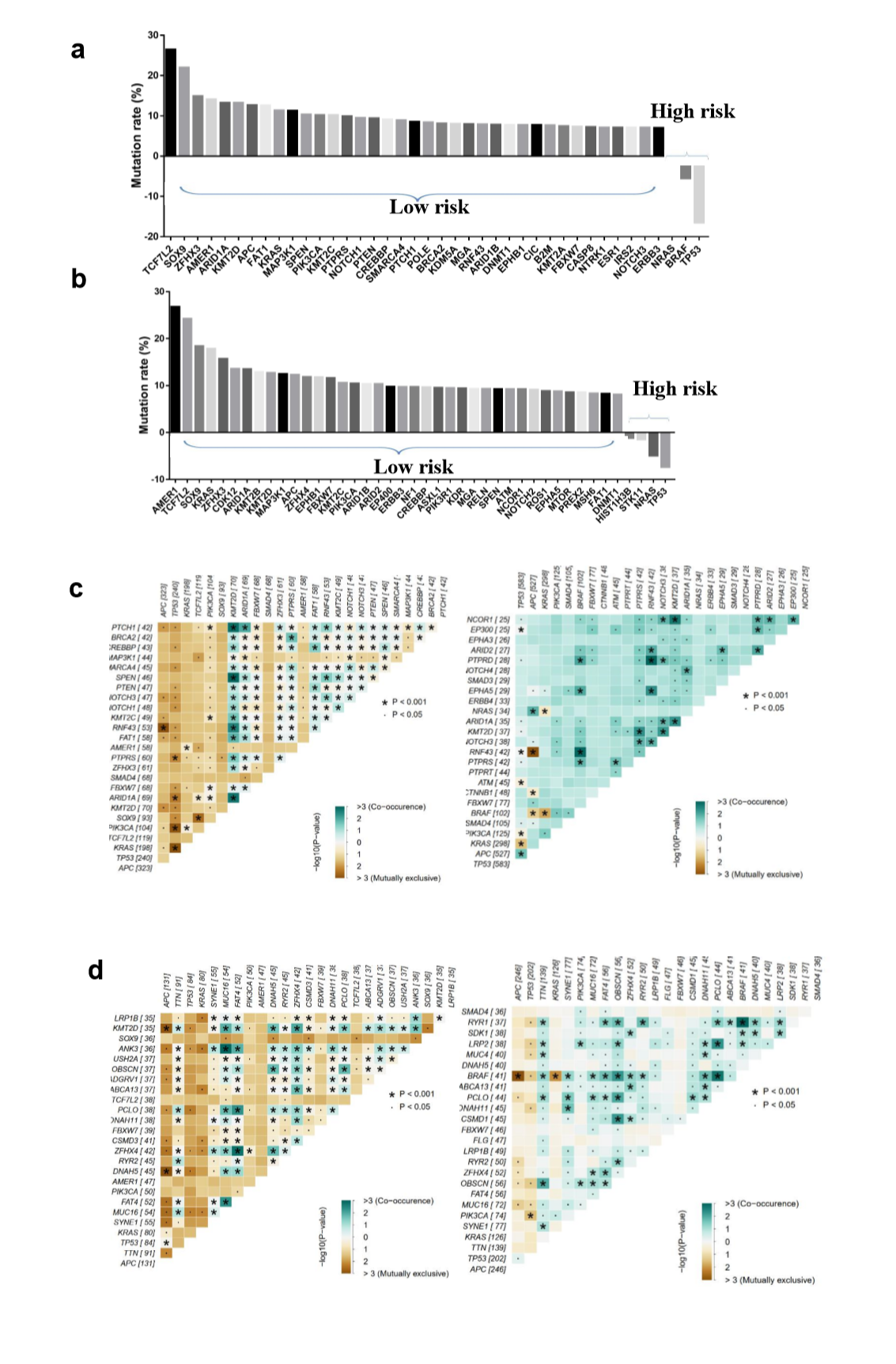
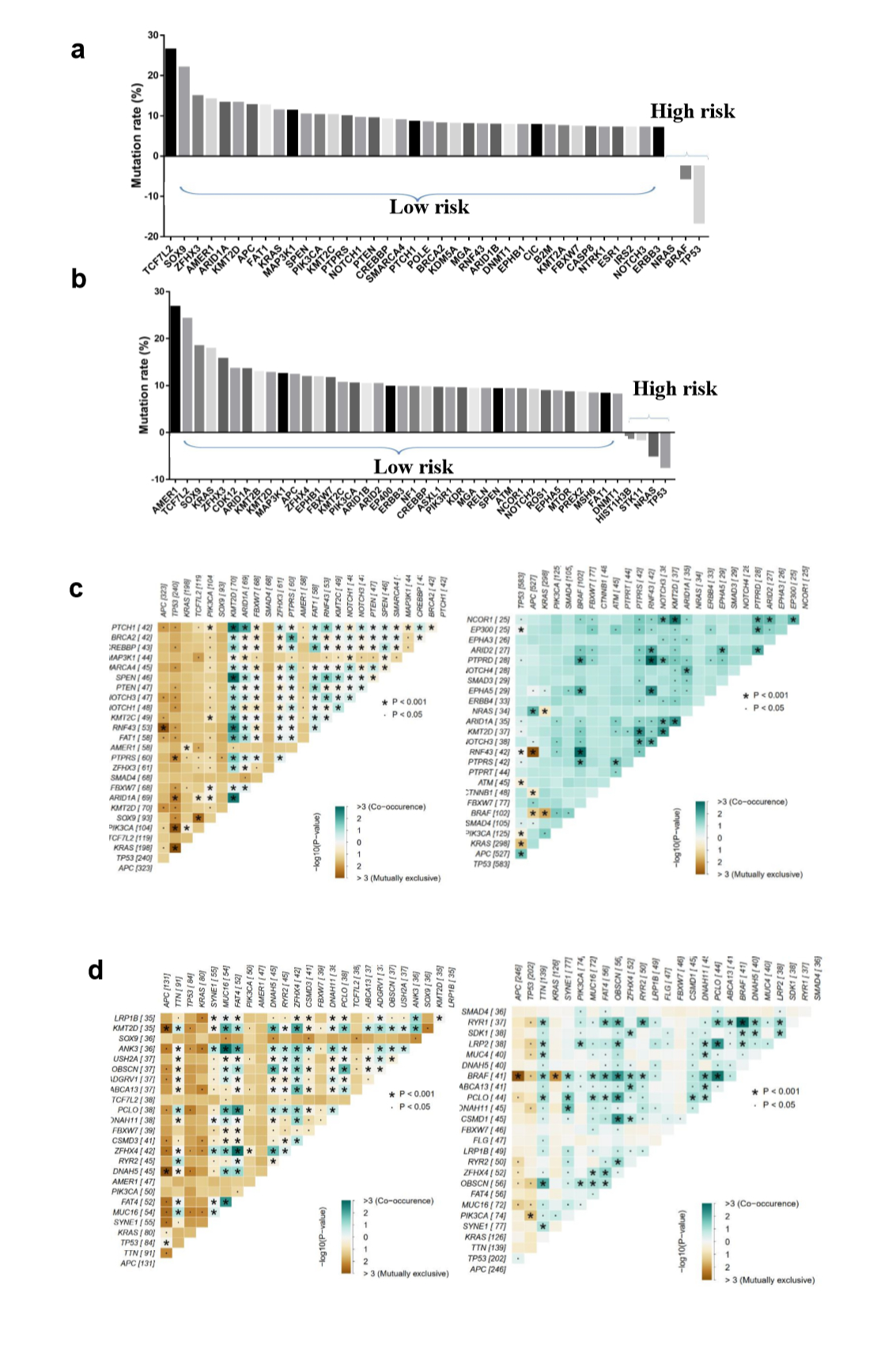
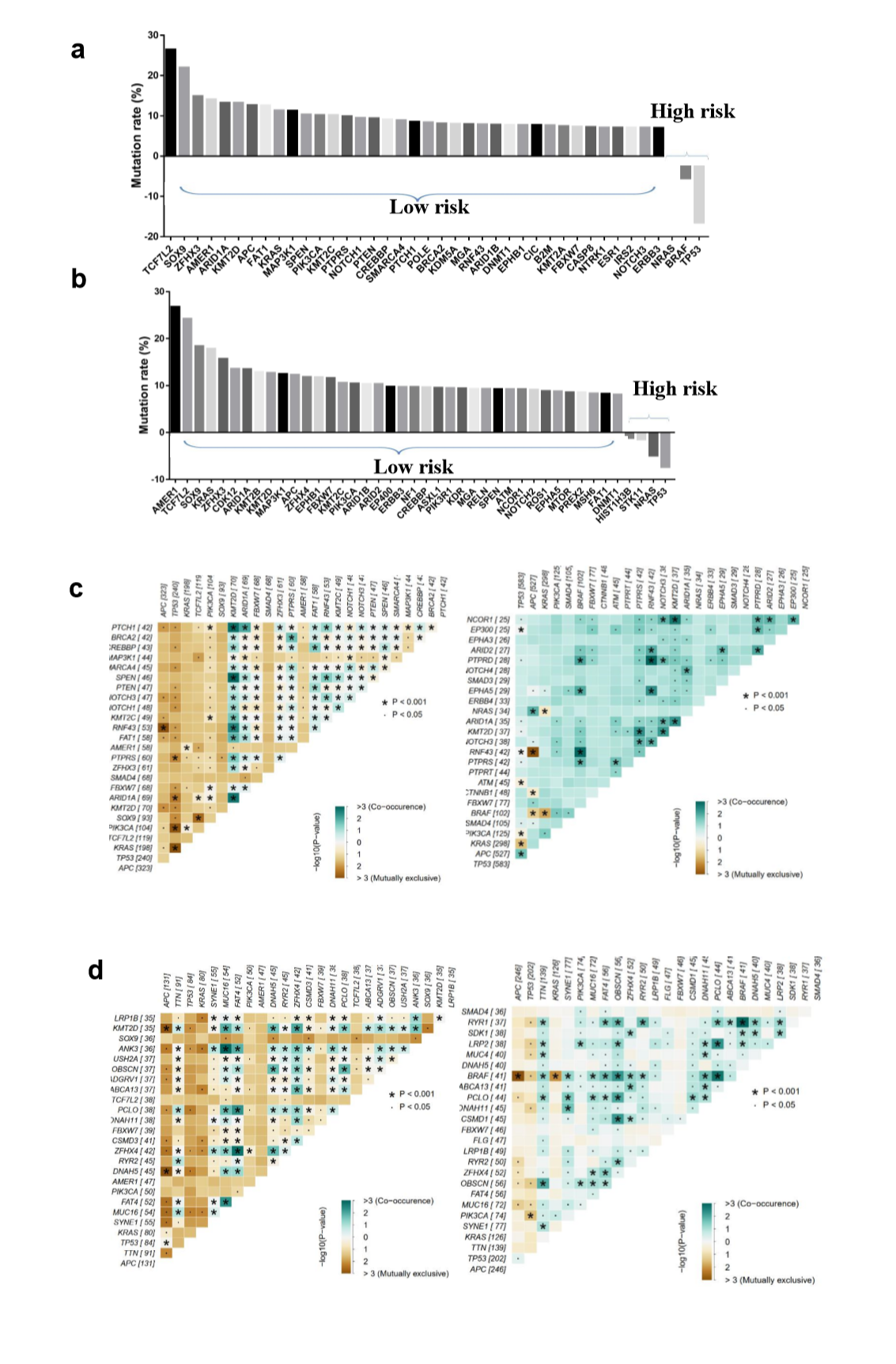
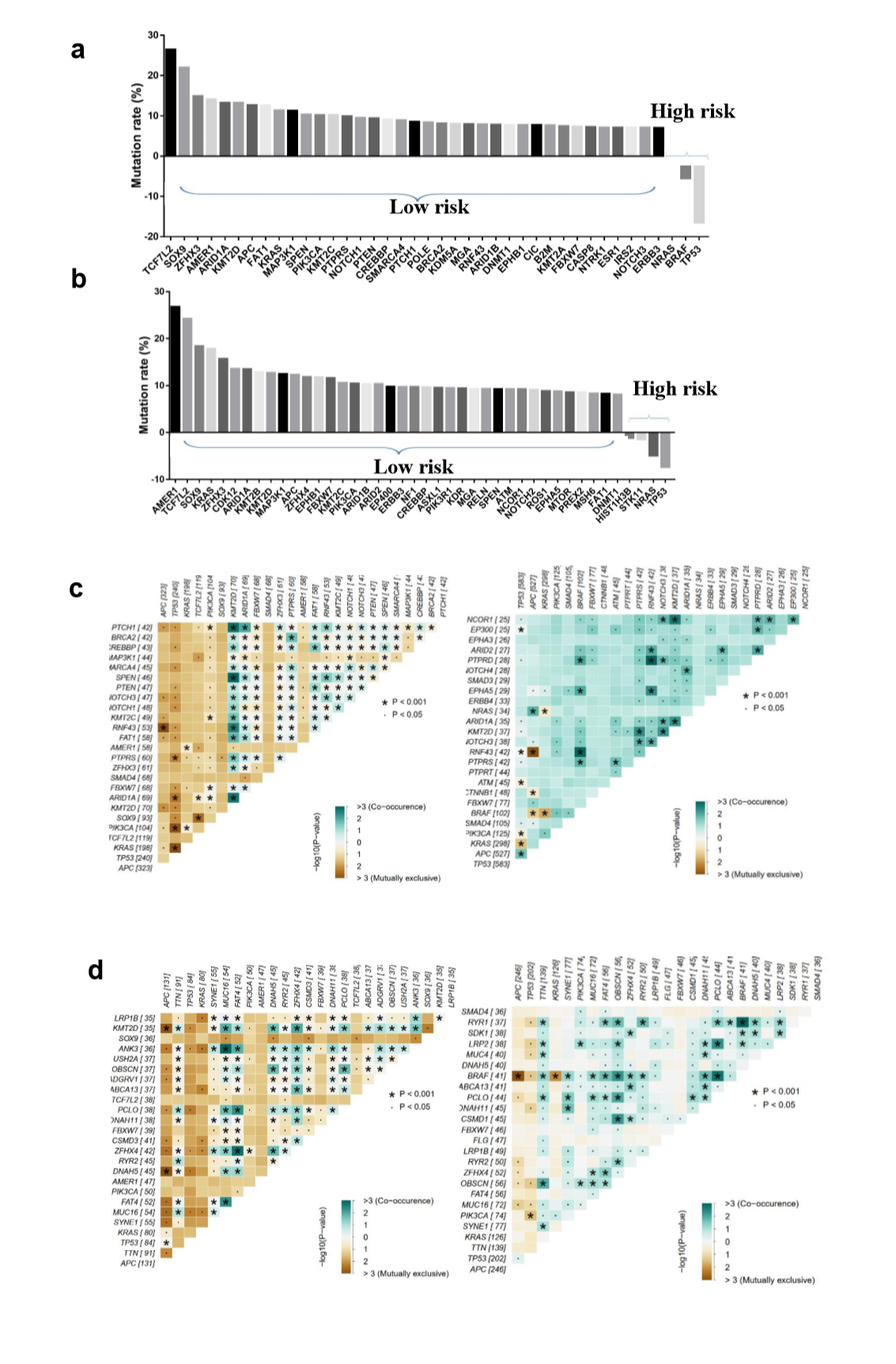
To explore the differences of genomic alterations between the defined high- and low-risk groups, we analyzed the data containing somatic mutations from TCGA (https://portal.gdc.cancer.gov/) database. First, comparison according to mutation frequency revealed a significant enrichment of different mutations between high- and low-risk groups (Fig. 4A and E). The most frequently found mutation types were missense mutations, nonsense mutations and frameshift deletions. Analyzing the mutation frequency of both subgroups, a larger number of mutations were found in the low-risk group as compared with the high-risk group (Fig. 4A). More than 95% genes had a higher mutation rate in the low-risk group as compared with the high-risk group (Supplementary Fig. S2A and B). In addition, the high- and low-risk groups had a significant different distribution of the top 10 mutated genes (Fig. 4C and G). These results suggested that there were significant differences in the mutated genes between the high- and low-risk groups.
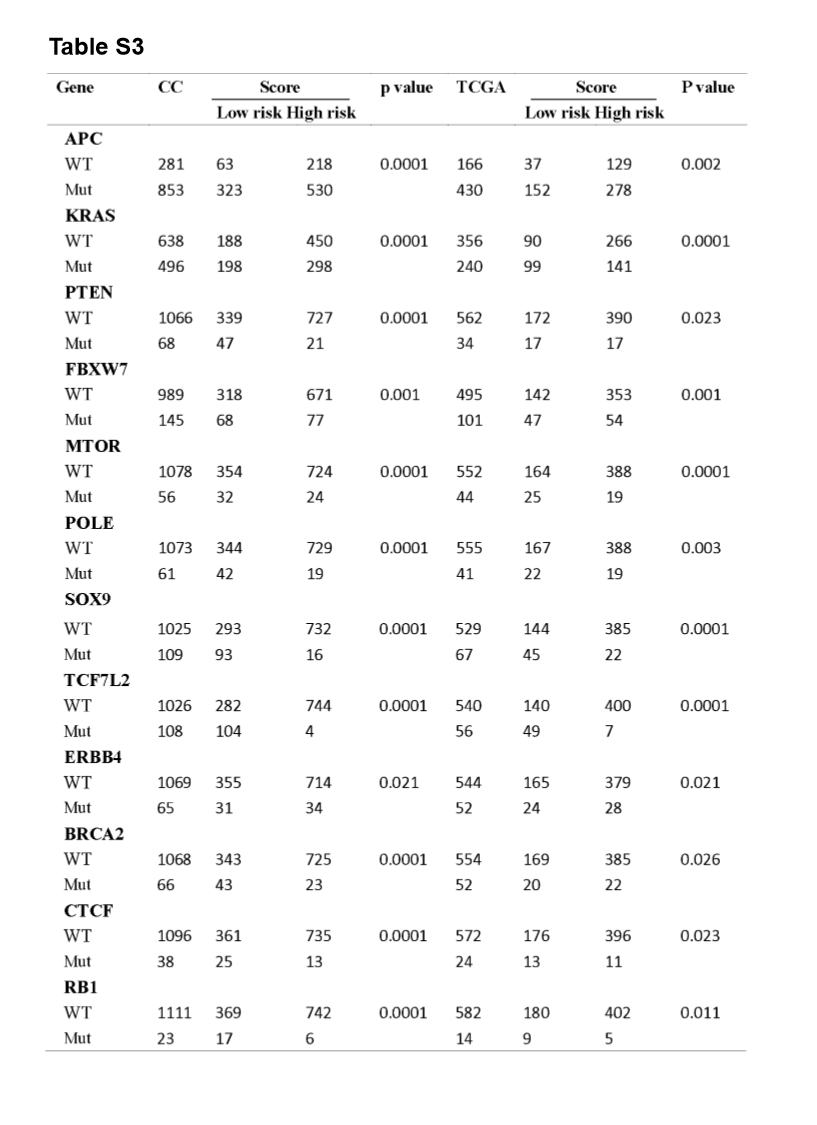
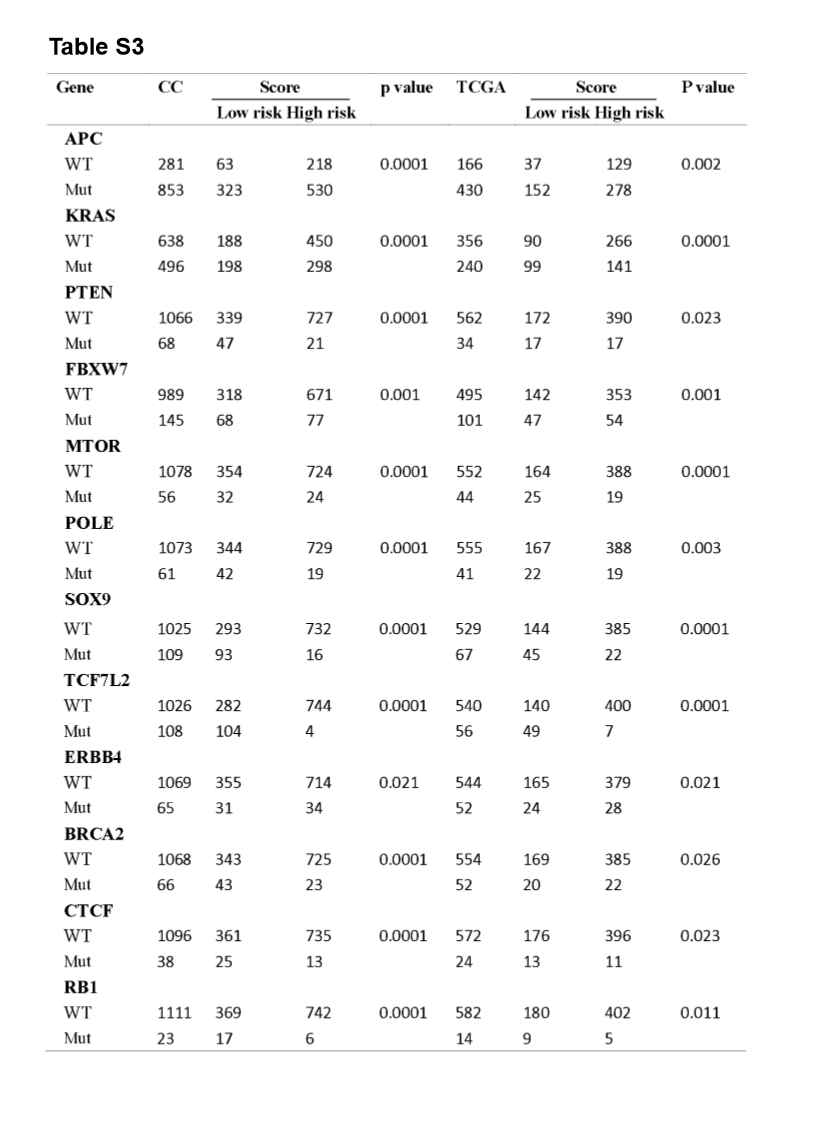
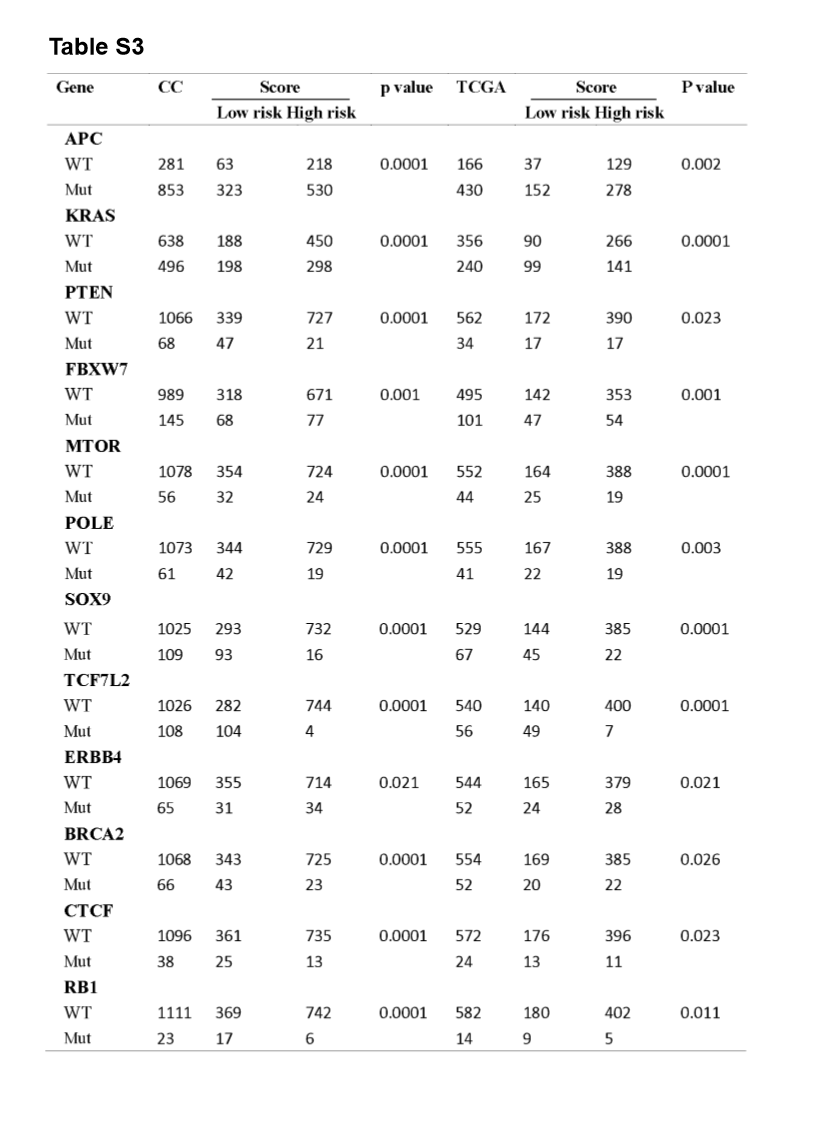
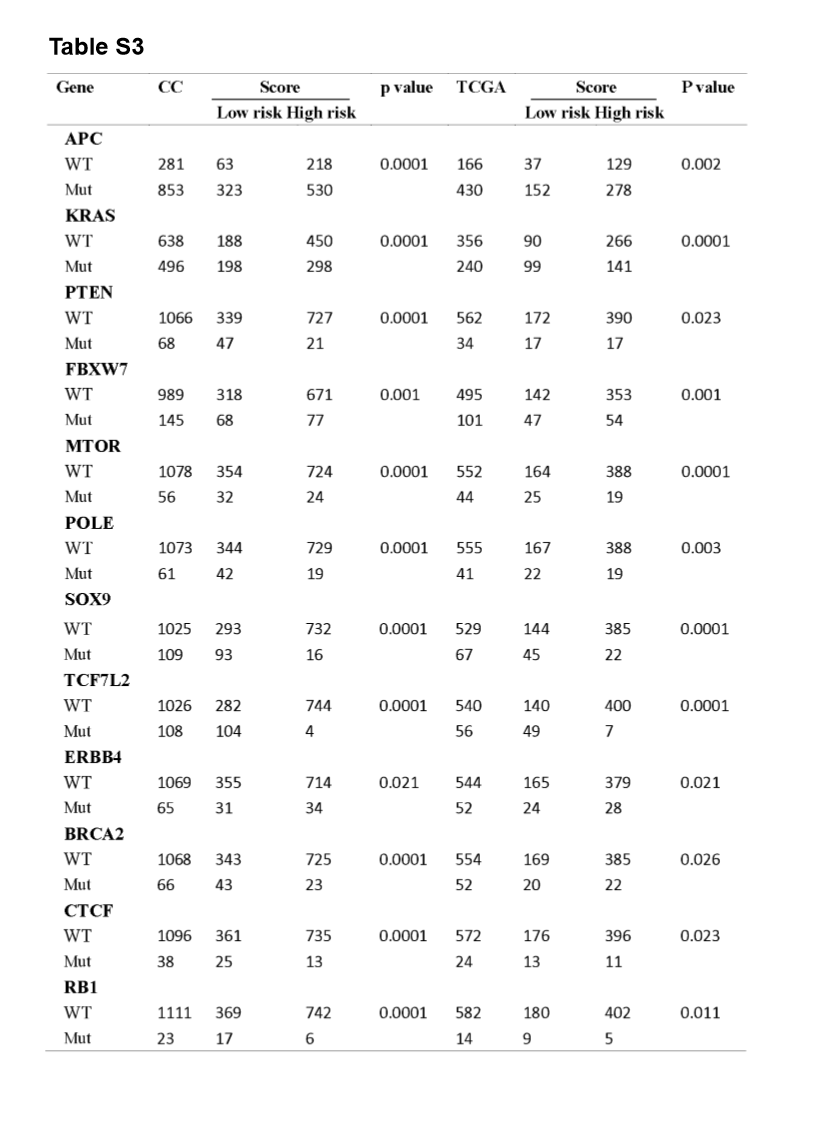
A significant enrichment of oncogenic alterations in such genes as BRAF, ZFHX3, SOX9 and MTOR were found in right-sided primary tumors, while oncogenic alteration of APC was primary found in the left-sided primary tumors. Analyzing mutated genes in MSI and MSS CRC patients, it revealed a higher altered frequency of genes including BRAF, TCF7L2, ZFHX3, MTOR and DNMT1 in MSI patient group as compared with MSS patient group, though a significant enrichment of oncogenic alteration in APC gene was found in MSS patients (Fig. 4D and H).
We observed significantly higher TMB in the low-risk group as compared with the high-risk group. Since mutational signatures are significantly correlated with TMB, which is positively correlated with tumor immune signatures and immunotherapy response, it can be speculated that mutational signature may be related to tumor immune activity and further affect immunotherapy response.
The mutational signature was associated with the genomic features of MSI and dMMR in CRC.
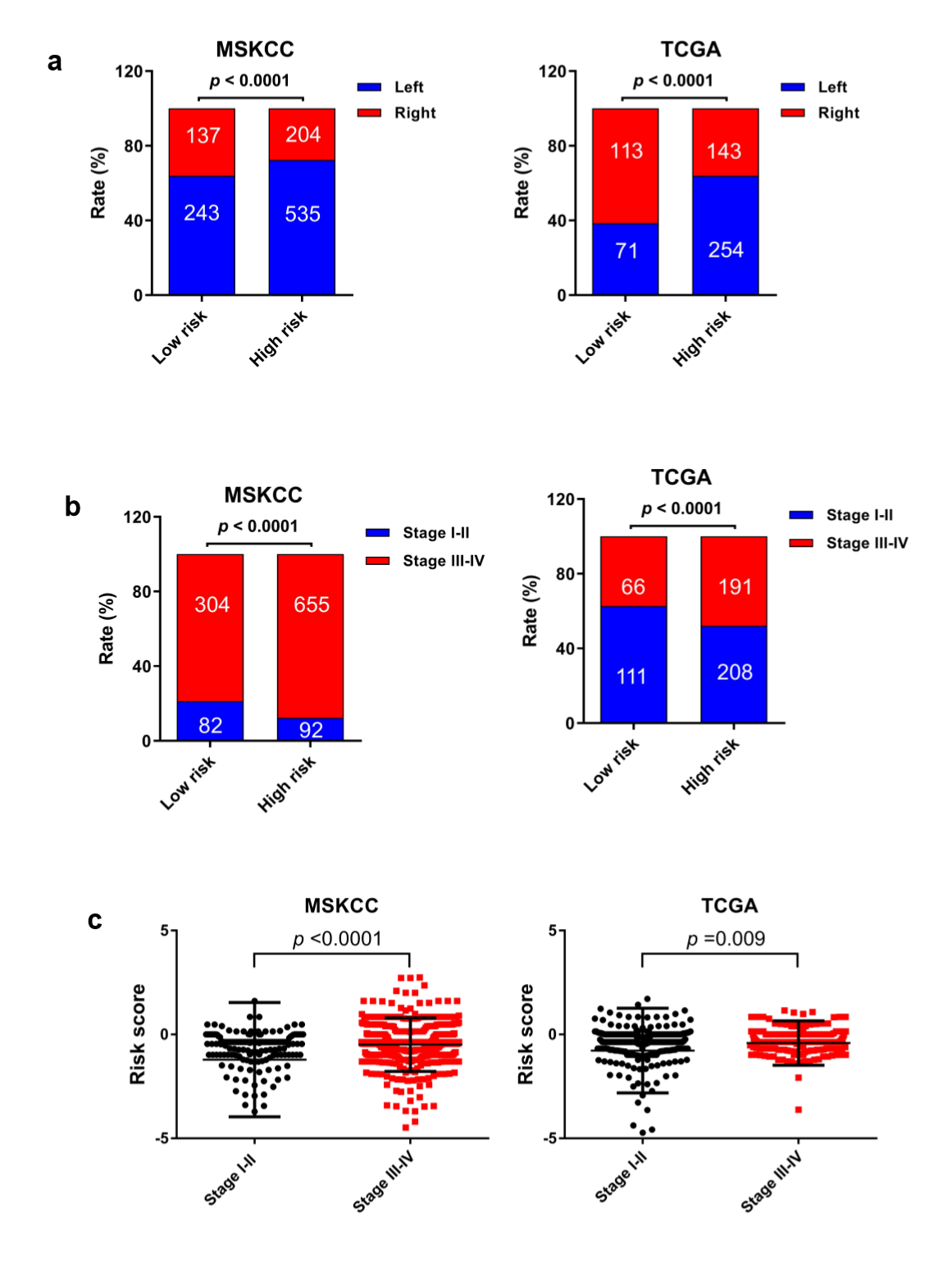
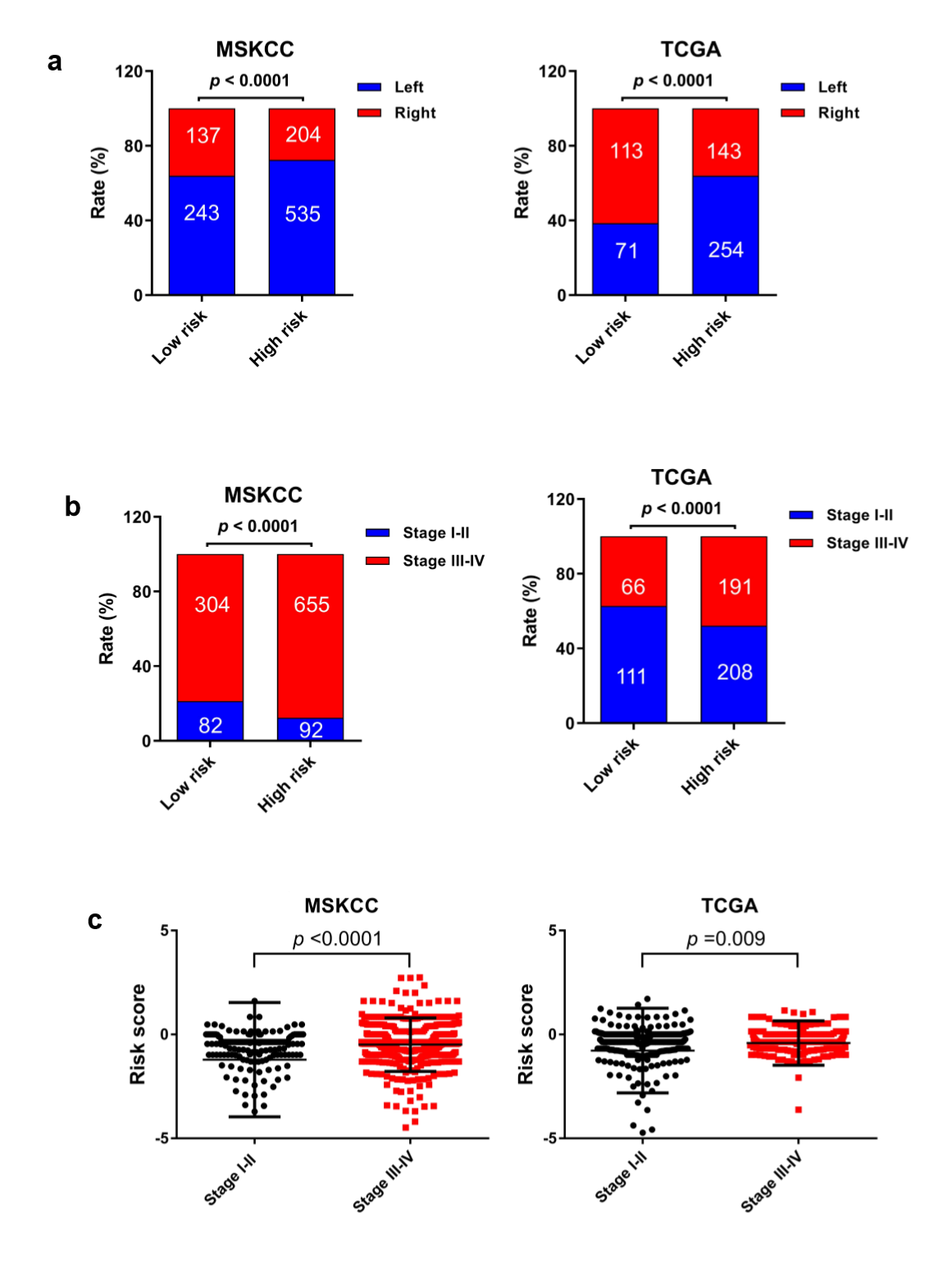
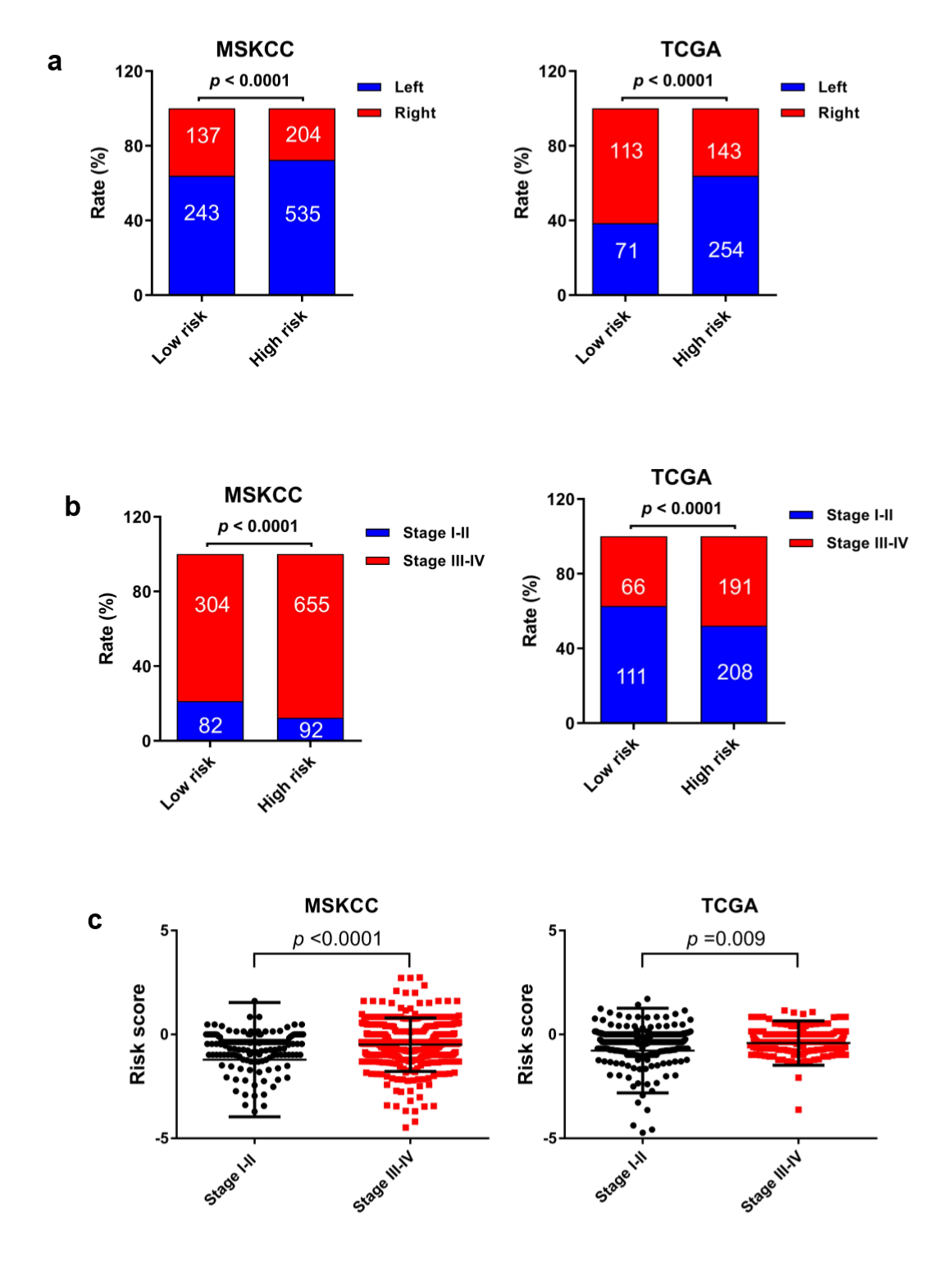
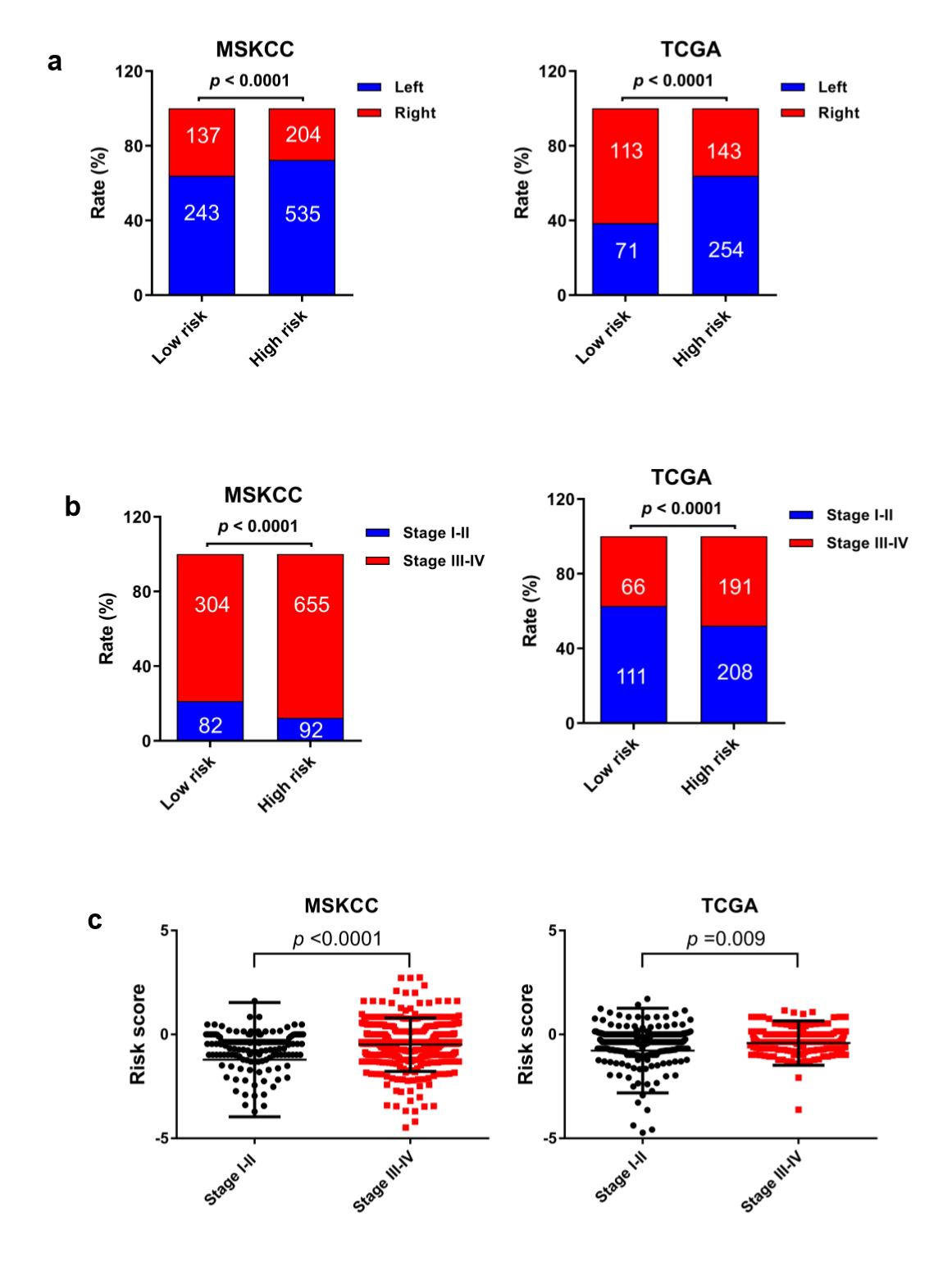
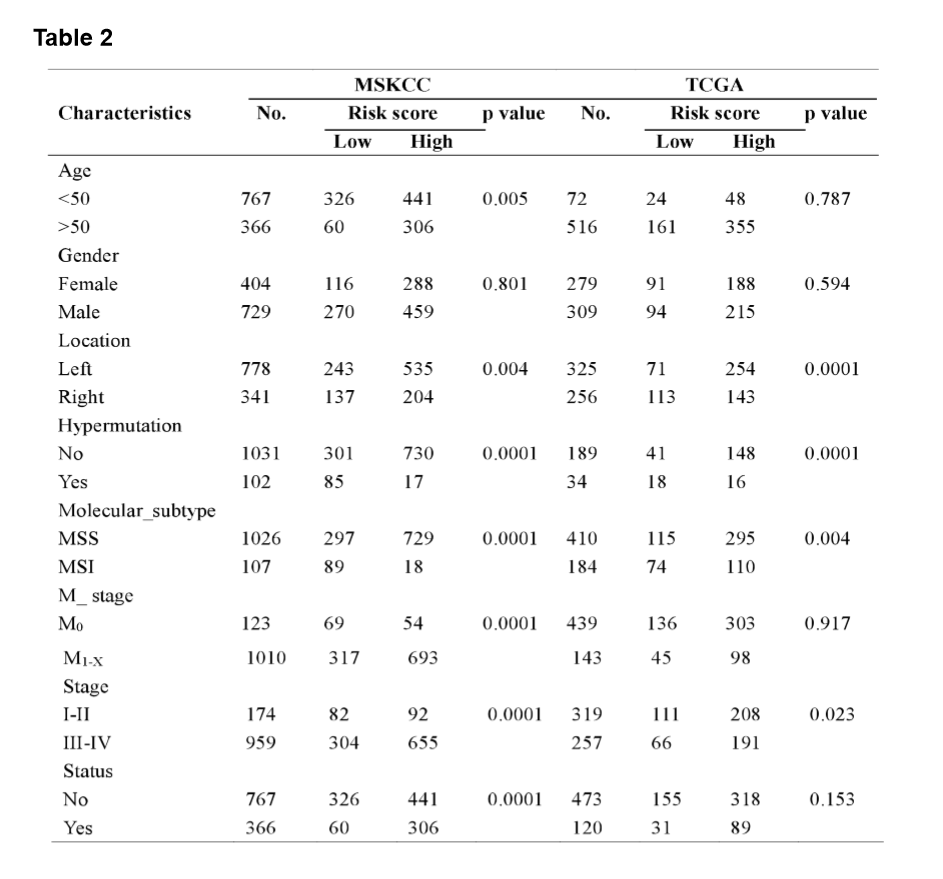
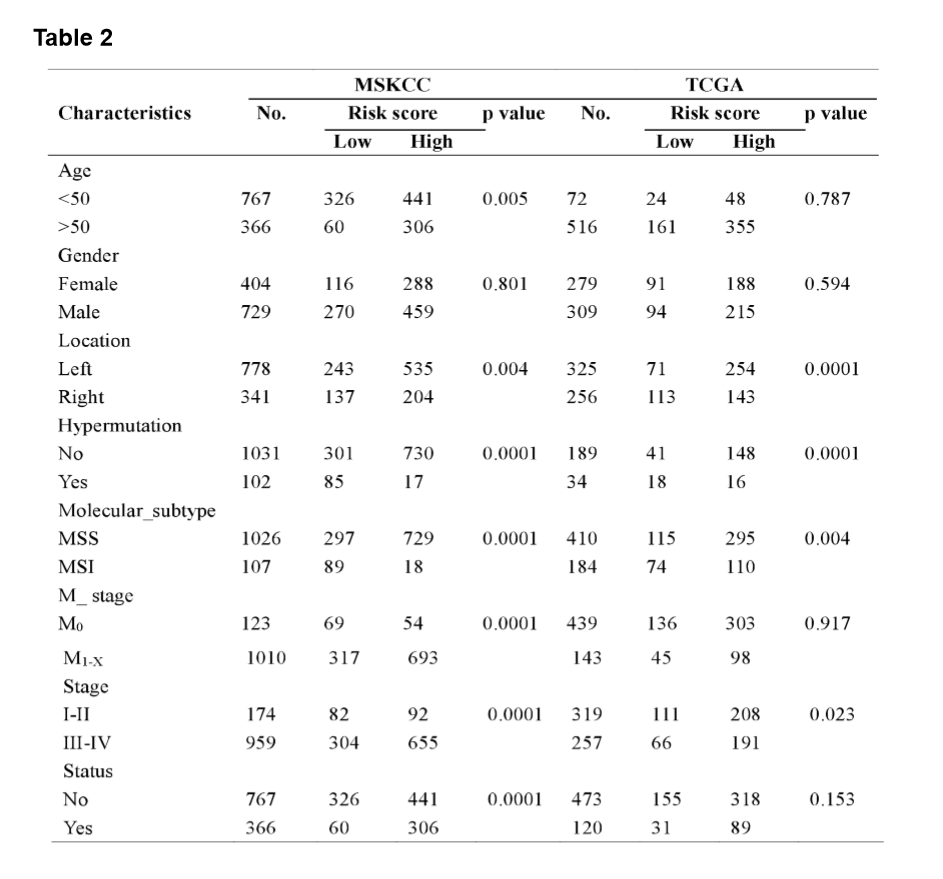
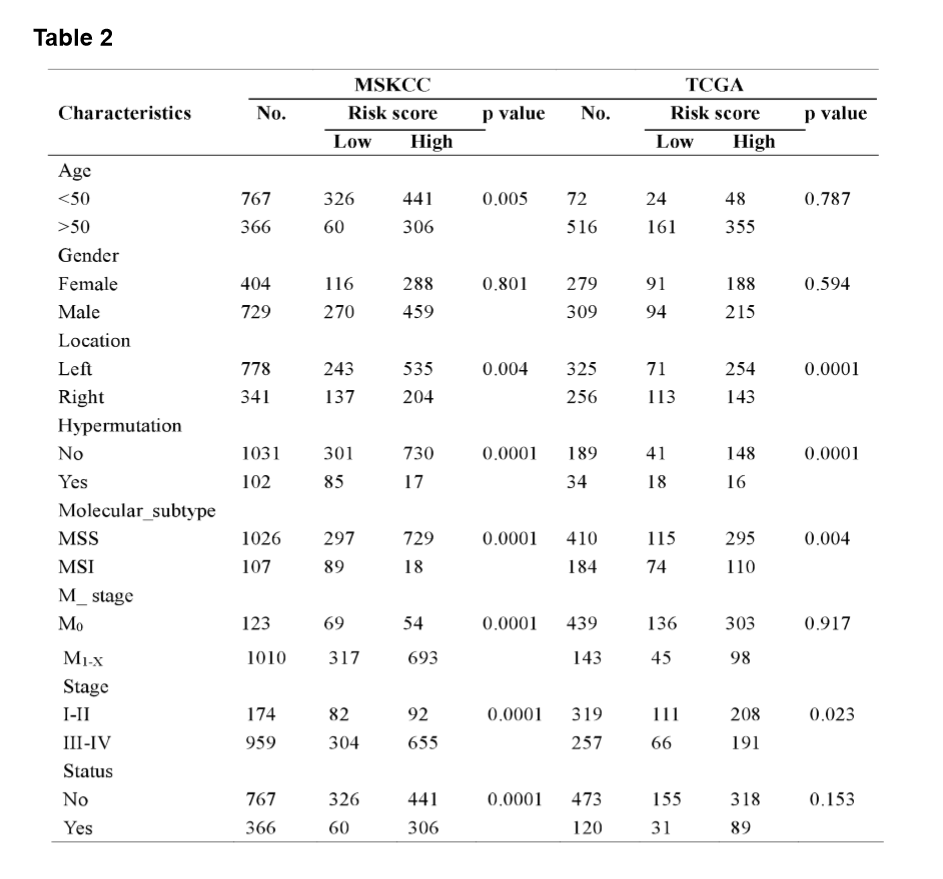
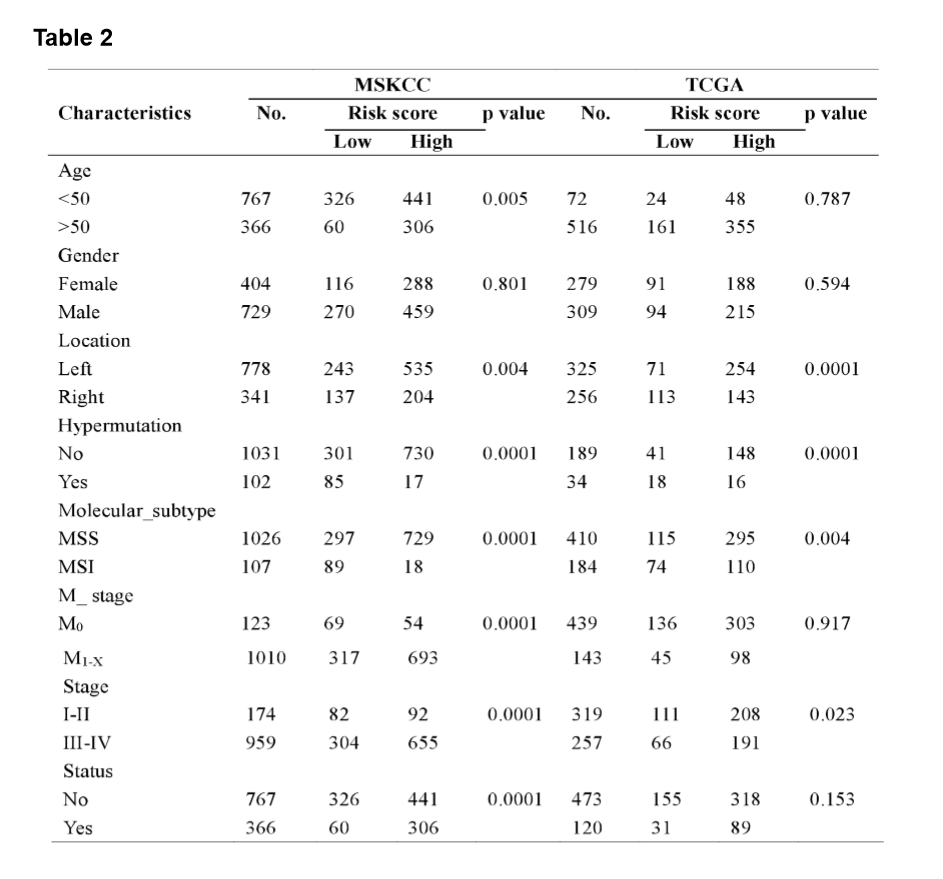
The MSI status is critical when considering immunotherapy and chemotherapeutic drugs as options for CRC patients [30]. MMR is the process by which potentially mutagenic misincorporation errors that occur during normal DNA replication are corrected and the absence of MMR results in increased accumulation of mutations [31, 32]. To further characterize the classified risk groups, we examined the association between the defined risk groups and other clinical characteristics using data of patients from both training and validation cohorts. The result showed that the outcome of the risk score was highly correlated with tumor location, hyper-mutation, MSI status, and TNM stage (Table 2). The risk score was observed to be significantly associated with the status of MSI/dMMR in both training and validation cohorts (Fig. 5A and B). In line with previous observation, the status of MSI was more common in low-risk group as compared with high-risk group (Fig. 5C). In addition, left-side tumors and TNM stage III-IV were more common in the high-risk group compared with the low-risk group (Supplementary Fig. 1A and B).
In TNM stage III-IV patients, the risk score was much higher than in that of patients with TNM stage I-II (Supplementary Fig. S1C). In addition, the defined low-risk group was significantly associated with hyper-mutation (Fig. 5D). Furthermore, we observed that high-risk group exhibited significantly higher rate of low mutation than low-risk group (Fig. 5E), and the number of mutations was higher in the low-risk group than that in the high-risk group (Fig. 5F). These results showed the mutational signature was associated with TMB, MSI status, and dMMR in CRC. It demonstrated a potential value of this mutation signature model for characterization of immune environment and prediction of immunotherapy outcome.
Mutation signature was associated with immune activity by immunogenic profiling identification.
To further clarify the relationship between mutational signature and immune-phenotyping, we analyzed single nucleotide variation (SNV) and transcriptome data in TCGA database. Immune activity differences between the high- and low-risk groups were determined by analyzing 29 immune-associated gene sets, which represented diverse immune cell types, functions, and pathways [33]. These gene sets were analyzed using the single sample gene set enrichment analysis (ssGSEA), an extension of GSEA, which could calculate separate enrichment scores for each pairing of a sample and gene set to quantify the activity or enrichment levels of immune cells, functions, or pathways in cancer samples [34].
On the basis of ssGSEA scores, we hierarchically clustered all CRC samples in TCGA dataset, and defined the three clusters as Immunity-High, Immunity-Medium, and Immunity-Low. Tumor purity, stromal score, and immune score were analyzed for each CRC sample based on the Estimation of Stromal and Immune cells in Malignant Tumor tissues using Expression data (ESTIMATE) algorithm. A heatmap of the infiltration levels and scores of each sample of immune cells in the three subtypes was shown in Fig. 6A. The results showed that the stromal score was significantly higher in the Immunity-High cluster and significantly lower in the Immunity-Low cluster. Immunity scores and ESTIMATE scores were gradually reduced from Immunity-H cluster to Immunity-L cluster. However, the opposite trend was observed for tumor purity in comparisons of the three CRC subtypes (Fig. 6A). Principal component analysis (PCA) revealed marked differences between the three clusters (Fig. 6B), indicating that Immunity-H cluster contained the largest number of immune cells and stromal cells, while Immunity-L cluster contained the largest number of tumor cells (Fig. 6D). Furthermore, we found significant higher expression of most human leukocyte antigen (HLA) genes in Immunity-H cluster as compared with Immunity-L cluster (Fig. 6C). Moreover, a significantly higher expression level of PD-L1 gene was found in Immunity-H cluster while Immunity-H cluster was correlated with better survival outcome as compared with Immunity-L cluster (Fig. 6E and 6F). According to distribution of these three clusters, we found that Immunity-H cluster was significantly enriched in the low-risk group (Fig. 6G), indicating that patients in the low-risk group might benefit more from PD-L1 inhibitor treatment.
To assess whether risk score was highly correlated with the immunity, we performed consensus molecular subgroups (CMS) classification [35], which give a more profound biological insight into metastatic CRC carcinogenesis, immunity typing, and has a strong prognostic effect. CMS1 is defined by an upregulation of immune genes and is highly associated with microsatellite instability (MSI-h) [36], while CMS4 is defined by an activated tissue growth factor (TGF)-β pathway and by epithelial-mesenchymal transition (EMT) making it in general more chemo-resistant. As expected, patients were divided into four clusters (Fig. 6H), and the distribution analysis revealed that the CMS4 was significantly more representable for the high-risk group as compared to CMS1, while the low-risk group was more represented by CMS1 subgroup (Fig. 6J). In addition, a significantly higher expression level of PD-L1 gene was found in the CMS1 subgroup as compared with other CMS subgroups (Fig. 6I). Taken together, the data suggested that the low-risk group was mainly represented by CMS1 and had a higher PD-L1 expression, which might benefit more from PD-L1 inhibitor treatment.
Composition of immune cell profiles in the high- and low-risk groups.
To further investigate the potential predictive value of the mutational signature for the immune status, we examined possible associations between the risk score and immune status. The risk score was negatively correlated with the TMB score (Fig. 7A) while TMB was positively correlated with immune score (Fig. 7B). Therefore, we postulated that the risk score was negatively associated with immune score. Since a high immune score was related with a better survival outcome (Fig. 7C), the low-risk group may also be associated with a better survival.
To Fig. out the infiltrated immune cell composition in the defined risk groups, we analyzed the expression signature matrix of 22 infiltrated immune cell types in tumor samples from the TCGA cohort using the CIBERSORT test. Among the total samples, 63 low-risk and 63 high-risk samples were found to be eligible for further analysis. The different immune cell fractions were weakly correlated with each other in tumor tissues in the TCGA cohort (Fig. 7D and 7E). Regarding to tumor-infiltrating immune cells in CRC microenvironment, reduced number of activated CD4 + memory T cells, but increased number of macrophages M0 were found in the high-risk group (Fig. 7F) as compared with the low-risk group. Finally, we analyzed the pathways that were significantly enriched in the high- and low-risk groups and found an enriched immunologic pathway in the high-risk samples (Fig. 7G). Taken together, these data suggested that mutational signature consisted of genes that are important regulatory components of the immune cell activation mechanisms. The predictive power of the mutational signature for OS might be dependent on the immune status of TME.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}