Study design and human Subjects. Patient samples were collected at Stanford University CA, USA, and the University of Heidelberg, Germany. Relapsing remitting MS (RRMS) was diagnosed according to the current McDonald criteria44,45. None of the patients met the diagnostic criteria for NMOSD, in particular spinal lesions spanning ≥3 segments46. Patients were tested for antibodies against aquaporin-4 and MOG and showed negative results. All included patients had elevated CSF white blood cell counts ≥10 cells/µl, and blood contaminated CSF samples were excluded by visual and microscopic inspection. Paired peripheral blood and CSF samples were obtained at the time of clinical onset (clinically isolated syndrome, CIS) or during an acute relapse. All but one patient had not received any MS-specific treatment prior to sample collection (Extended Data Table 1). All experimental protocols were approved by the institutional review board of Stanford University (IRB# 34529) and the ethics committee of the medical faculty of the University of Heidelberg (IRB# S-466/2015). Written informed consent was obtained from each patient.
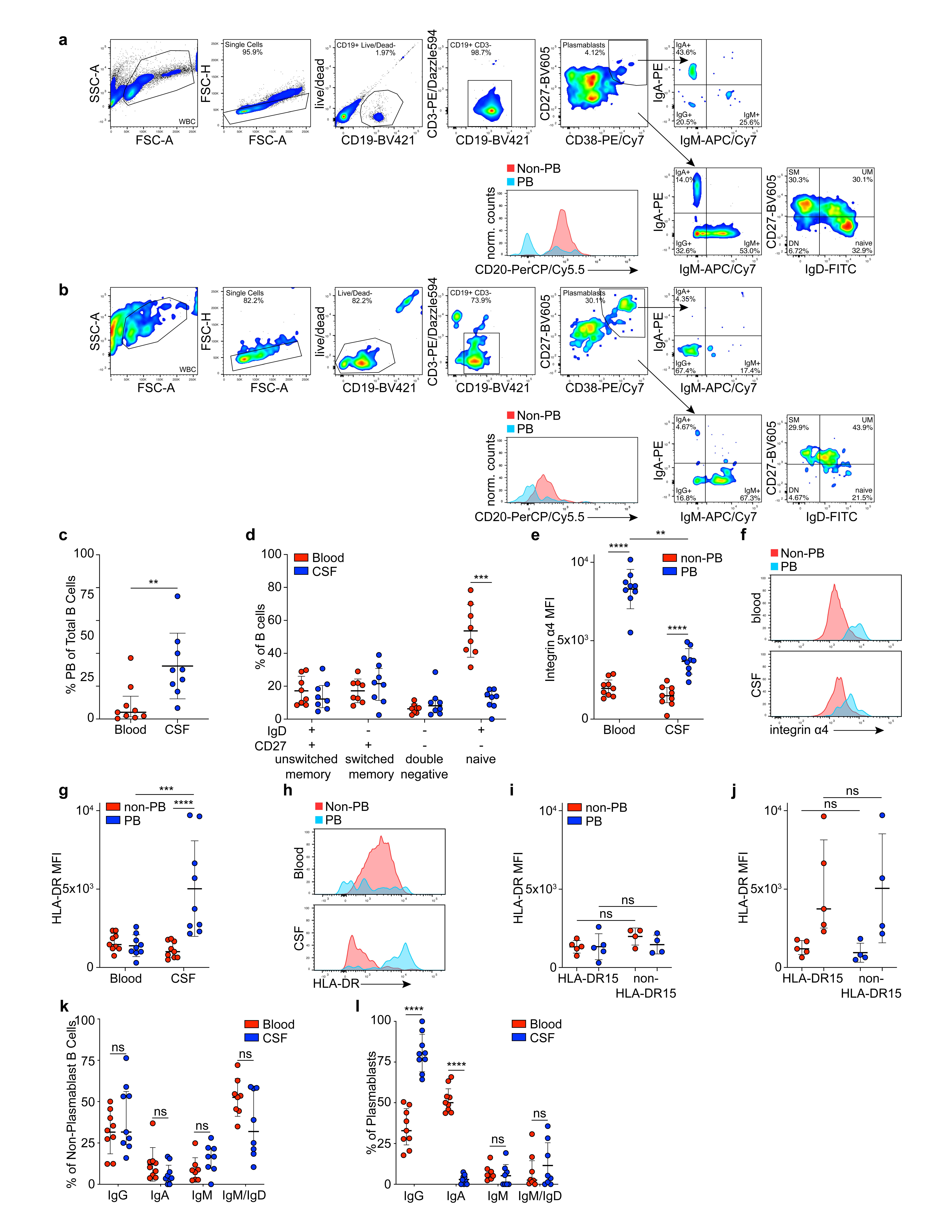
Cell preparation, antibody staining, and flow cytometric cell sorting. CSF was centrifuged immediately after lumbar puncture and cells were counted. PBMCs were isolated from heparin blood by density gradient centrifugation using Ficoll PLUS media (Cytiva). Cells were magnetically separated with anti-CD19 magnetic beads (Dynabeads CD19 Pan B cell isolation kit, Invitrogen), then stained according to standard protocols, using antibodies against the following cell surface markers: CD20 (clone L27, dilution 1:10), CD38 (clone HB7, dilution 1:30), IgD (clone IA6-2, dilution 1:20) (all BD Biosciences), CD3 (clone OKT3, dilution 1:60), CD19 (clone HIB19, dilution 1:20), CD27 (clone O323, dilution 1:20), IgM (clone MHM-88, dilution 1:40), HLA-DR (clone L243, dilution 1:100), α4 integrin (clone 9F10, dilution 1:100) (all BioLegend), IgA (clone IS11-8E10, dilution 1:20) (Miltenyi Biotec), and Sytox blue (ThermoFisher Scientific, dilution 1:500). Single cells were sorted with a FACSAria II cell sorter (BD Biosciences) using FACSDiva (v8.0, BD Biosciences) into 96-well PCR plates (BioRad). For single-cell repertoire sequencing, plasmablasts were sorted from PBMC (plasmablast gate (4.12%) in panel 5 in the representative flow cytometry plots shown in Extended Data Fig. 1a). All B cells were sorted from CSF (B cell gate (73.9%) in panel 4 in the representative flow cytometry plots shown in Extended Data Fig. 1b). FlowJo Version 10.7.1 (BD) and R version 3.6.1 was used to evaluate flow cytometry data.
Single-cell BCR repertoire sequencing. BCR repertoire sequencing was carried out using our in house developed plate-bound single-cell sequencing technology as described previously15,47,48. Briefly, reverse transcription with oligo-dT was carried out in separate wells, attaching unique well-ID barcodes by template switching activity of Maxima Reverse Transcriptase (ThermoFisher Scientific). Barcoded cDNA from each plate were pooled and amplified in 3 consecutive PCRs, including attaching plate-specific barcodes and sequencing adapters. PCRs were carried out separately for HC of IgG, IgA, and IgM, as well as for κ for λ LC, and separate libraries were generated from each, gel-purified, cleaned with Ampure XP beads (Beckman Coulter), and sequenced on an Illumina MiSeq (Illumina) with 2 x 330 paired-end reads.
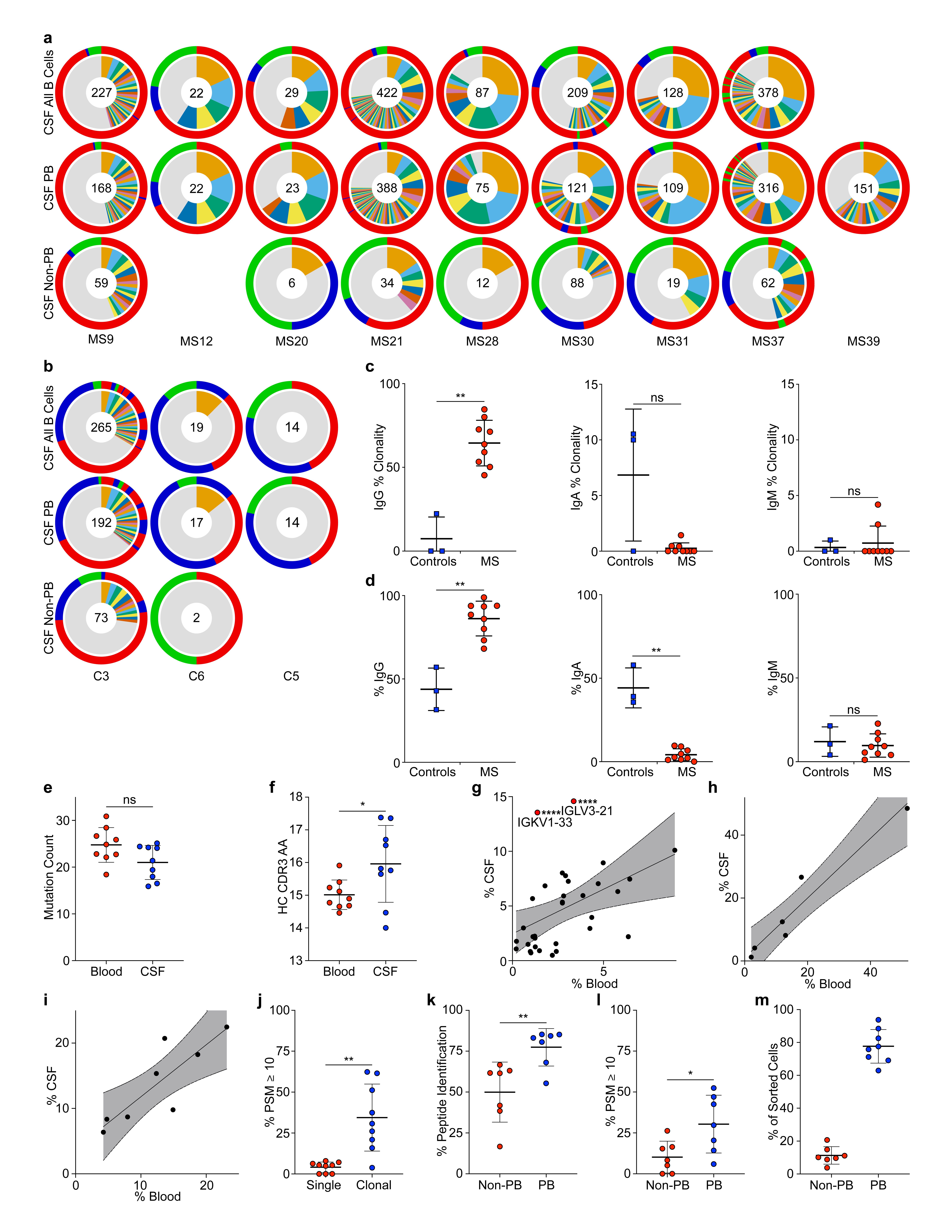
Sequence analysis. The MiSeq FASTQ workflow was used for Fastq generation and plate demultiplexing. R version 3.6.1 was used for custom analyses. Paired reads of sequences that passed quality thresholds were stitched and separated by plate and well IDs. Similar reads sharing the same plate and well IDs were clustered into operational taxonomic units (OTUs)49. Consensus sequences were aligned to germline variable-chain immunoglobulin sequences with IMGT HighV-QUEST v1.3.150, which reports V, D, and J germline genes, HC and LC CDR3-lengths, and non-silent mutation counts and locations. Clonal expansions were defined based on sharing the same HC and LC V and J genes and exhibiting >70% amino acid identity within the HC and LC CDR3s. Percent clonality represents the percent of all sequences that fulfill these criteria. To calculate IGHV, IGLV, IGHJ, and IGLJ gene usages, percent abundance of each particular gene was calculated in blood and CSF PB of each patient and means were calculated across patients. Genes that were present in less than three CSF samples were excluded from this analysis. While our sequencing method preferentially captures PB sequences due to higher amounts of immunoglobulin mRNA (Extended Data Fig. 2m), enough non-PB B cell sequences passed filter thresholds to compare the non-PB repertoire to the PB repertoire in 7 patients (Extended Data Fig. 2a,b). For patient samples MS12 and C6 only PB were captured (while gating on all B cells), and for MS39 only PB were sorted. For phylogenetic analysis, sequences were binned according to their HC V-gene family and V-gene. Concatenated LC and HC were then aligned with Muscle51 and clustered with PhyML52 using maximum-likelihood clustering. Each tree-partition was rooted by their HC V-gene. Phylogenetic trees were drawn in Python using the ETE 3 toolkit53.
Peptide identification with mass spectrometry. Immunoglobulins were purified from 1.5ml of CSF samples with Protein A (ThermoFisher Scientific). The purified IgGs were reduced with 0.02 M dithiothreitol at 57°C for 1 hour, alkylated with 0.05 M iodoacetamide at room temperature (RT) in the dark, and digested with trypsin overnight at RT. Peptides were extracted and desalted as previously described54. An aliquot of the peptide mixtures was loaded onto an Acclaim PepMap 100 precolumn (75μm × 2cm, C18, 3μm, 100Å) in-line with an EASY-Spray, PepMap column (75μm × 50cm, C18, 2μm, 100Å) with a 5μm emitter using the autosampler of an EASY-nLC 1000 (ThermoFisher Scientific). The peptides were gradient eluted into a Lumos Fusion Tribrid (ThermoFisher Scientific) mass spectrometer using a 120min gradient from 5% to 35% solvent B (90% acetonitrile, 0.5% acetic acid), followed by 10 minutes from 35% to 45% solvent B and 10 min from 45 to 100% B. High resolution full MS spectra were acquired with a resolution of 120,000, an AGC target of 4x105, a maximum ion time of 50 ms, and scan range of 400 to 1800 m/z. Following each full MS scan as many data-dependent HCD MS/MS spectra were acquired in the orbitrap as possible in a 3 second cycle time. Monoisotopic precursor selection (MIPS) was set to peptide, precursors with a charge state of 2 – 7 and minimum intensity of 5x104 were selected for MS/MS. Dynamic exclusion was set to 60 seconds after a single selection. All MS/MS spectra were collected using the following instrument parameters: resolution of 30,000, an AGC target of 105, maximum ion time of 120 ms, two microscans, 1.6 m/z isolation window, and Normalized Collision Energy (NCE) of 32.
The MS/MS spectra were searched against the respective peptide specific database including common contaminant proteins using the search engine Byonic54. The search parameters were set to trypsin allowing two missed cleavages, fixed modification of carbamidomethyl on cysteine, variable modification of oxidation on methionine and deamidation on glutamine and asparagine. Peptides mapping to variable regions of IgG were manually verified. In order to include only sequence-specific peptides, peptides that aligned to non-immunoglobulin or constant-region sequences were excluded from the analysis, as were peptides that aligned to the repertoire of multiple patients. Included were peptides that aligned to one variable sequence in a single patient. Peptides that aligned to more than one variable sequence in a single patient were included if all matching sequences were exact matches or clonally related, in which case the peptide was counted as representative for all matches. Counts of identical or non-identical peptide spectral matches (PSM) per sequence were tallied for each sequence. Sequences that had >1 or >10 matching peptides were presented as percentage of all sequences (Fig. 1j,k, Extended Data Fig. 2g,h). The mass spectrometry files are accessible at MassIVE (massive.ucsd.edu) under accession number MSV000086829.
Selection and recombinant expression of mAbs. Representative antibodies from the largest clonal B cell expansions in the CSF of each patient were selected for recombinant expression. In patients with more than 10 large clonal expansions, sequences were preferentially chosen based on their usage of one of the 11 most abundant IGHV genes in the CSF (Extended Data Fig. 3). HC and LC variable sequences were custom generated (IDT), and cloned into pFuse vectors (Invivogen), containing human IgG constant region or kappa or lambda constant regions, respectively. Fab HC were expressed in in-house plasmids, containing the constant-region C1 up to Cys103. Plasmids were transfected into Expi293T cells using Expifectamine (ThermoFisher Scientific). Culture medium was harvested after 4 and 7 days post transfection. mAbs and Fabs were purified with protein A and protein G resins, respectively (ThermoFisher Scientific). Antibody concentrations were measured with a nanodrop spectrophotometer (ThermoFisher Scientific) and hIgG quantitation ELISAs (Bethyl Laboratories) and checked for purity on SDS protein gels with Coomassie staining.
Protein expression and purification. EBNA1 proteins and peptides were obtained from: full-length AA1-641 (Abcam), AA328-641 (Virion Serion), AA408-641 (ProspecBio). GlialCAM proteins and peptides: full-length AA34-416 (OriGene), ECD AA34-234 (Novoprotein), and ICD AA262-416 with N-terminal His-Tag was cloned into a pet30(+) vector, expressed in BL21 chemically competent E. coli (Sigma Aldrich) to an OD of 600nm, and induced with IPTG (Sigma Aldrich) for 3h at 37˚C. Cell pellets were disrupted by sonication and proteins were purified with cOmplete His-Tag Purification Resins (Roche Life Science), followed by size-exclusion purification (Cytiva). For all other used peptides and proteins see Supplementary Tables 2-4.
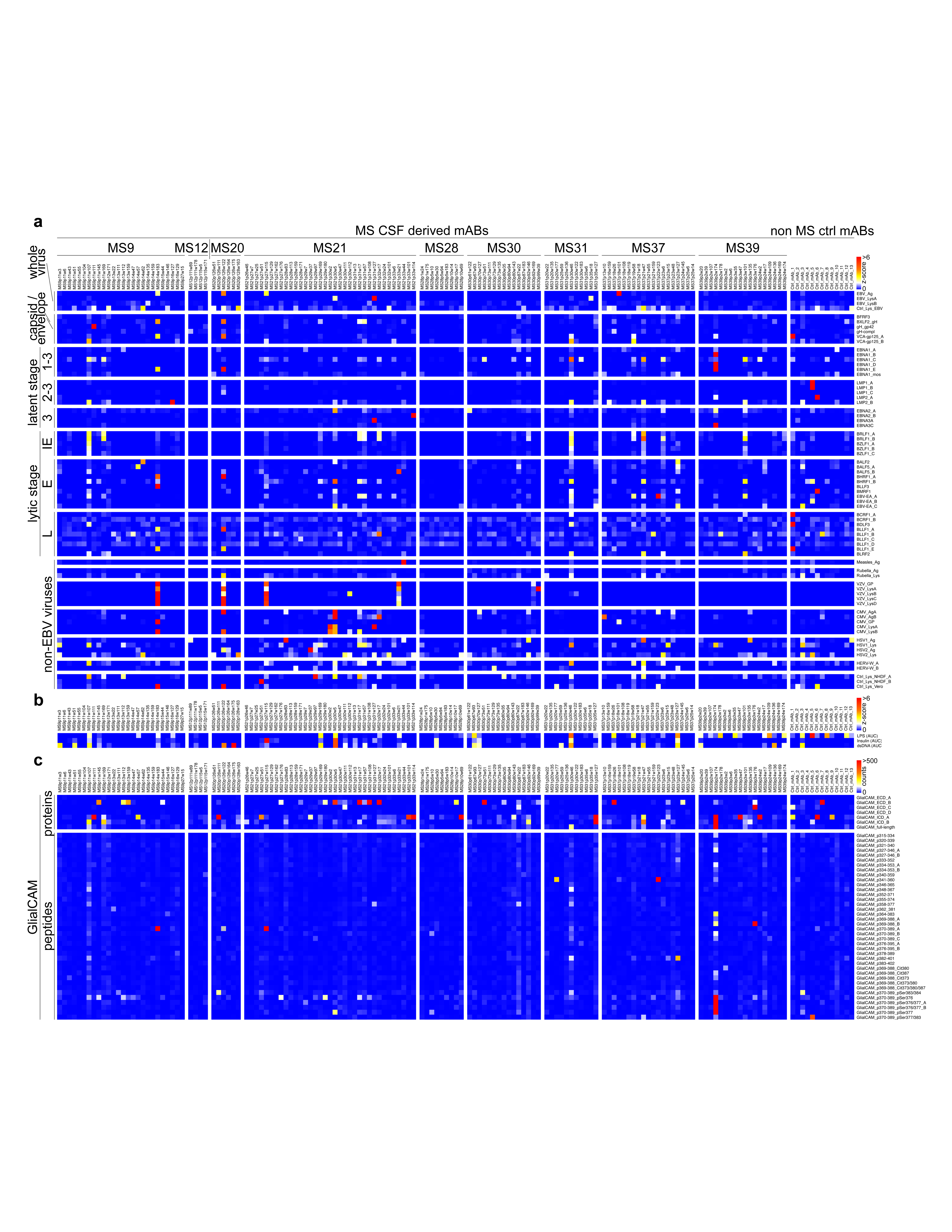
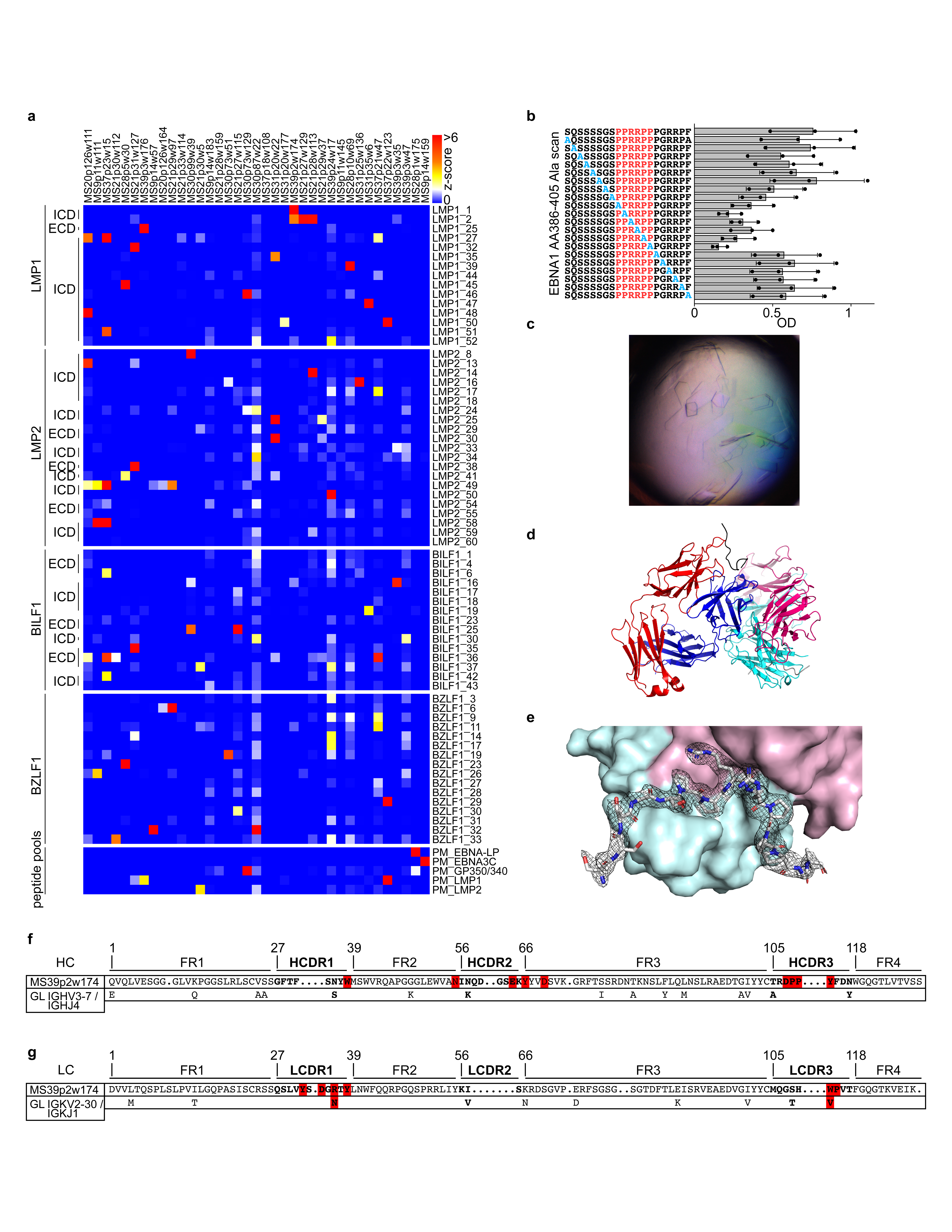
Planar protein microarrays. Protein microarrays were generated as described previously 55,56 (https://web.stanford.edu/group/antigenarrays/). In brief, peptides, recombinant proteins, and lysates were diluted at the indicated concentrations in a 1:1 solution of PBS/water and protein printing buffer (ArrayIt) (Supplementary Tables 2-4), aliquoted on 384-well plates, and printed on SuperEpoxy Slides using a NanoPrint LM210 system (ArrayIt). Two independent quadruplicates of each analyte were spotted, and some proteins were used in several versions / preparations from different sources (Supplementary Table 2). Ready-made HuProt Arrays version 3.1 were obtained from CDI labs. Arrays were circumscribed with a hydrophobic marker, blocked overnight at 4 °C in PBS containing 3% FCS and 0.1% Tween-20, and incubated with individual mAbs at a concentration of 1 µg/ml for 1h at 4˚C, then washed twice for 20 min in blocking buffer on a rotating shaker. Arrays were then incubated with Cy-3-conjugated secondary goat anti-human IgG (0.8 μg/mL) (Jackson ImmunoResearch) for 1h at 4 °C, then washed twice for 30 min in blocking buffer, twice for 30 min in PBS, and twice for 15 s in water. Arrays were spun dry and scanned with a GenePix 4000B scanner (Molecular Devices). Median pixel intensities for each fluorescent spot were determined with GenePix Pro-3.0 software (Molecular Devices). Z-scores for each row of antigens were calculated for viral antigens, raw intensities were analyzed for GlialCAM arrays. Heatmaps were generated with Morpheus software (The Broad Institute; https://software.broadinstitute.org/morpheus).
PhIP-Seq. PhIP-Seq was performed using a human proteome-wide library expressing overlapping 49-amino-acid peptides with a 24-AA sliding window approach starting at the N-terminus. Briefly, 2uL (1ug/mL) of substrate antibody was diluted 1:100 in blocking buffer for two sequential rounds of immunoprecipitation. After the second round of immunoprecipitation and amplification in E. coli, next generation sequencing libraries were prepared for paired-end 150 base next generation DNA sequencing on the Illumina Hi-Seq platform as previously described28,57. After alignment of the reads to the reference peptide sequences, quality control was performed and only reads present at an abundance of at least 10 reads per hundred thousand were carried forward. The number of reads mapping to each peptide were then counted and individually scored as a percentage of the total.
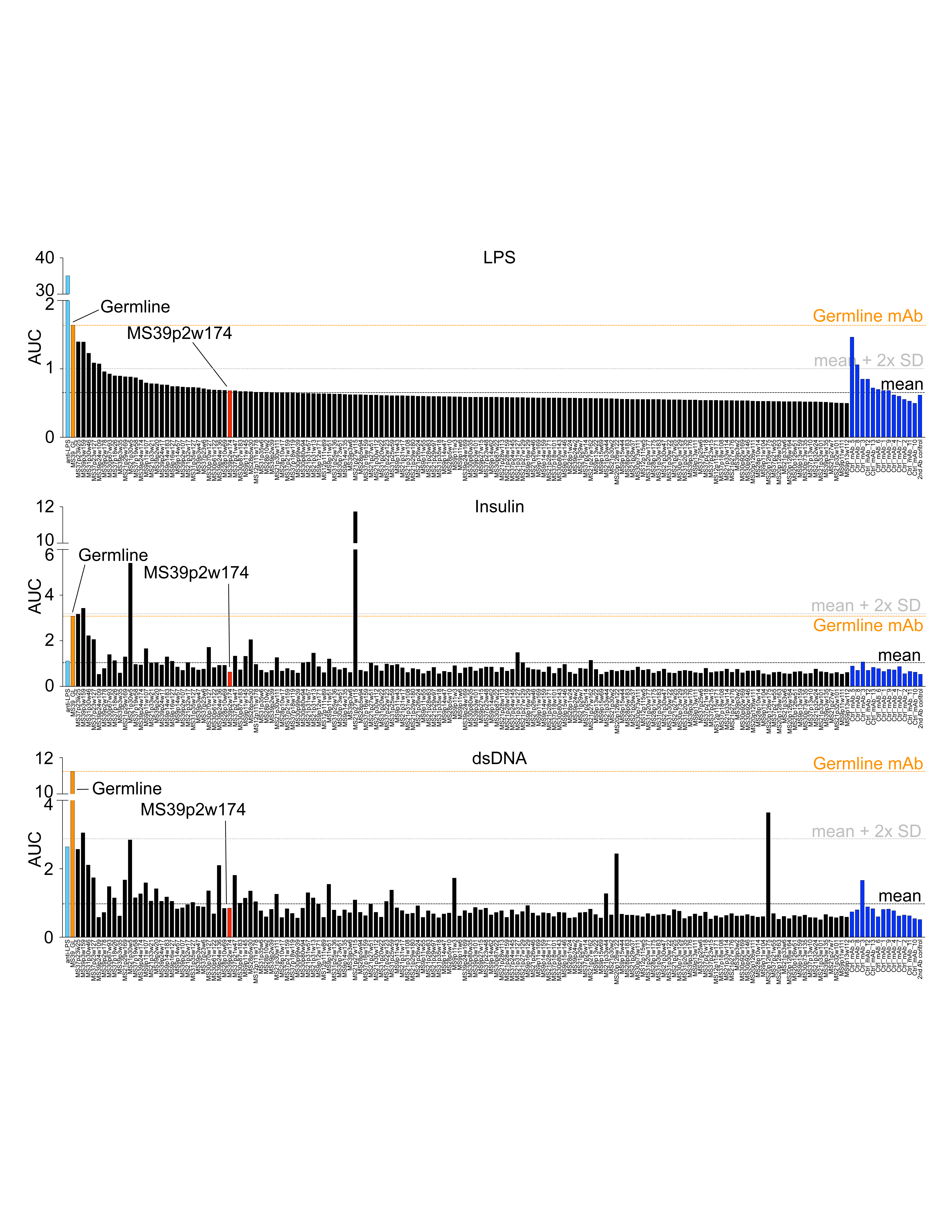
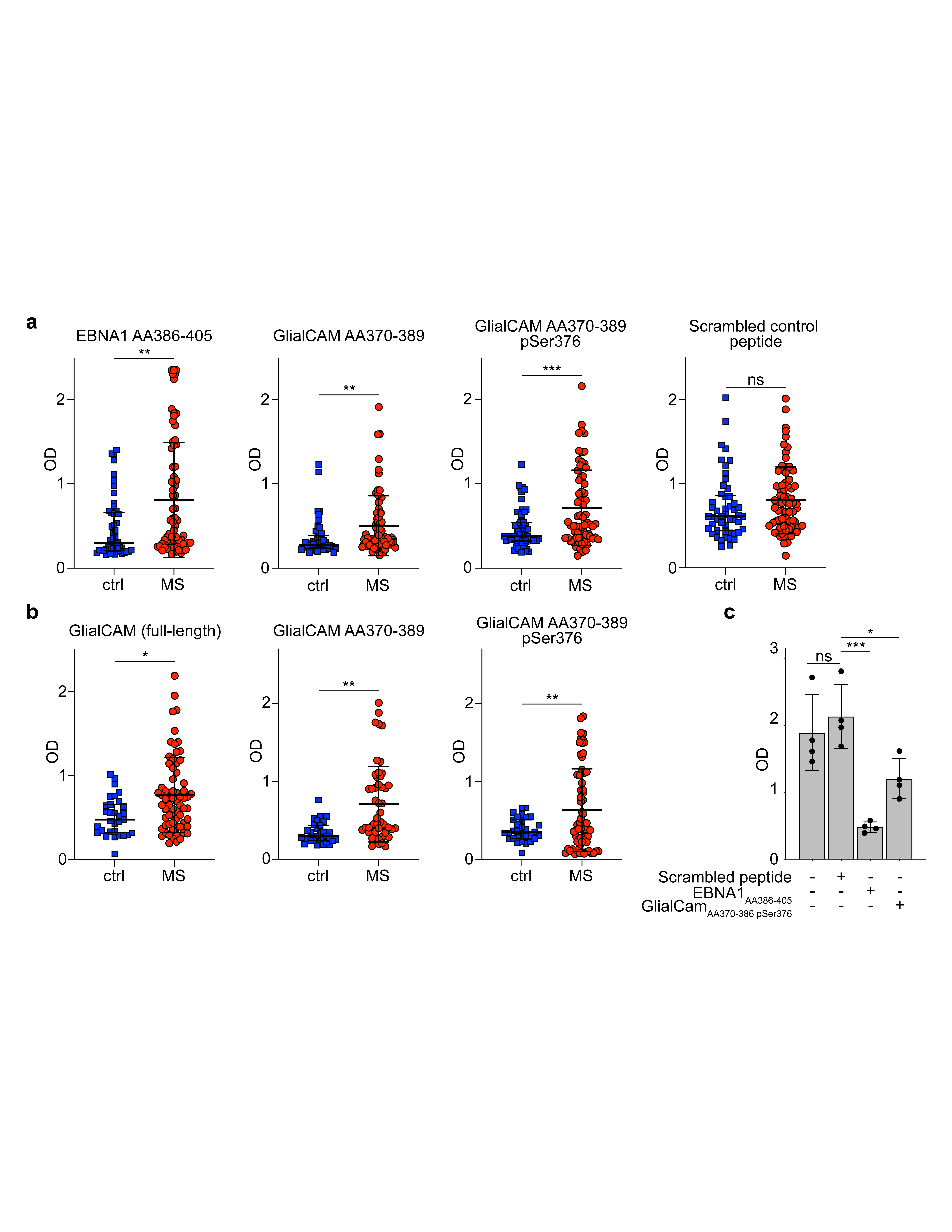
ELISA. Cytokine ELISA kits were used according to manufacturers’ instructions: mouse IL-6, IL-10, IL-12, IFN-γ, and TNF (BD Biosciences), and IL-17A (ThermoFisher Scientific). For protein and peptide ELISAs, MaxiSorp 384-well plates (ThermoFisher Scientific) were coated with 1 µg/ml peptide or protein in carbonate-bicarbonate buffer at 4˚C overnight, then washed 6x with PBST (PBS + 0.05 % Tween20), blocked with blocking buffer (PBS + 1% BSA) for 1h, and mAbs were applied at 1 µg/ml in blocking buffer. Human and mouse plasma samples were diluted 1:100 and T cell supernatants 1:4 in blocking buffer. After overnight incubation at 4˚C, plates were washed again 6x with PBST, secondary antibody HRP-conjugated goat anti-human IgG (Jackson ImmunoResearch), was applied for 1h at RT, and after 6 additional washes with PBST, plates were developed with TMB substrate (ThermoFisher Scientific), stopped with 1N sulfuric acid, and read on a SpectraMax Paradigm Microplate Reader (Molecular Devices). For plasma ELISAs with blocking of plasma IgG, MaxiSorp 384-well plates were coated with 2µg/ml recombinant Protein G (Acro Biosystems) at 4˚C over night, then washed 6x with PBST, and incubated with 1:100 diluted plasma at 4˚C over night. Plates were again washed 6x with PBST, and then incubated with the respective blocking peptides at 10µg/ml for 2h at RT. Then, biotinylated EBNA1AA385-405 was added at 1µg/ml and incubated for 1h at RT. Plates were washed again 6x with PBST, incubated with HRP-conjugated streptavidin (BioLegend) for 1h at RT, and developed with TMB substrate as described above.
Western blotting. Western blots were run according to standard protocols. Briefly, purified proteins were boiled in Laemmli-buffer with 10% beta-mercaptoethanol for 5 minutes, run on 4-12% Criterion XT Bis-Tris protein Gels (Bio-Rad), then transferred onto a nitrocellulose membrane using a Trans-Blot Turbo semi-dry transfer system (Bio-Rad) and stained with MS39p2w174 at 10µg/ml or with mouse anti-EBNA1 antibody (Biorbyt) or mouse anti-GlialCAM antibody (R&D Systems) followed by secondary HRP-conjugated goat anti-human IgG and anti-mouse IgG (Jackson ImmunoResearch). Coomassie gels were run concomitantly, fixed with 10% methanol and 7% acetic acid, and stained according to standard protocols. Uncropped western blot and Coomassie images are available in Supplementary Figure 1.
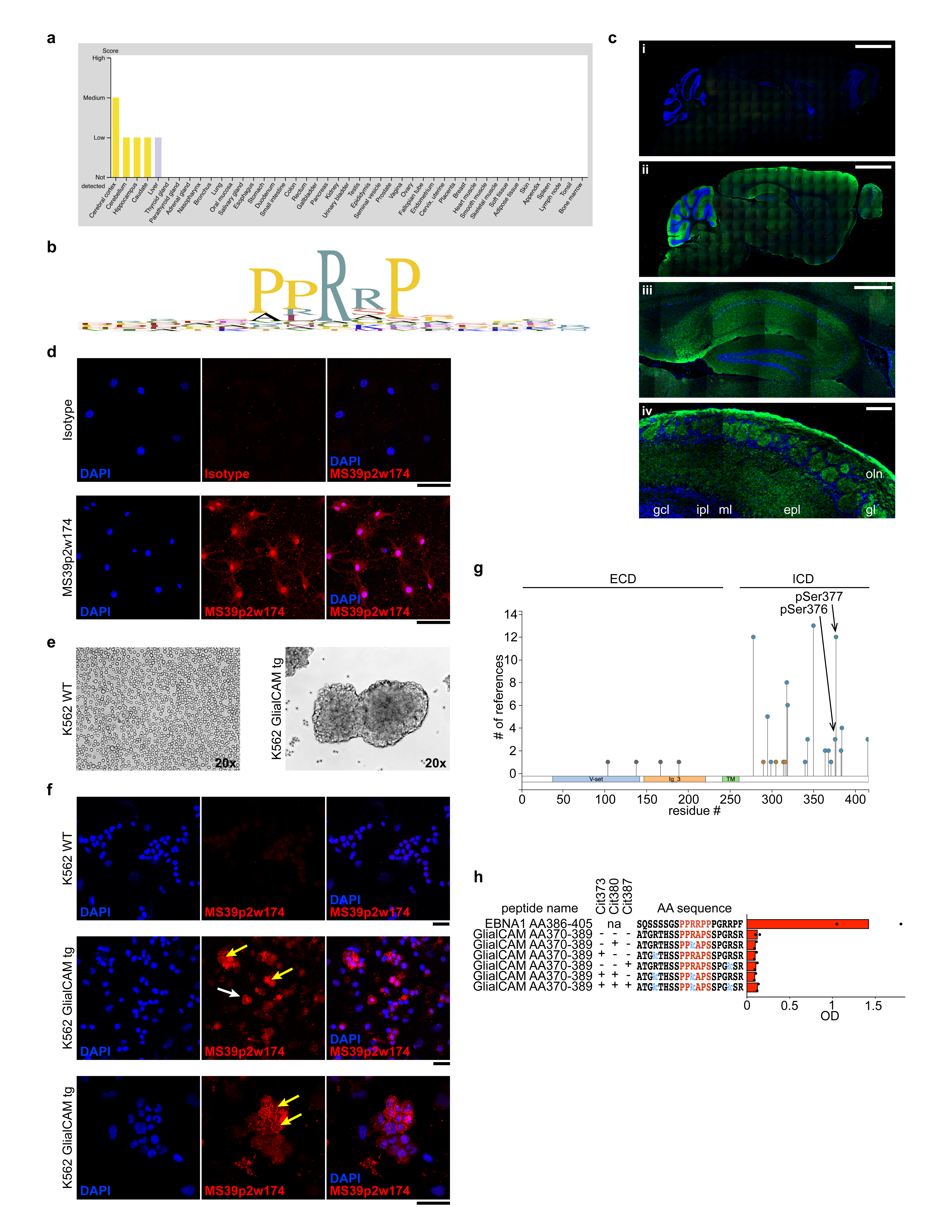
Fluorescent Immunohistochemistry on mouse brain slices and Immunofluorescence on primary cultured rat oligodendrocytes. An adult mouse (F1 generation of FVB x C57blk6 cross) was transcardially perfused with 4% paraformaldehyde and post-fixed in 4% PFA overnight at 4C. After sucrose equilibration, the brain was blocked in OCT and sectioned at 12µm on a standard cryostat. Sections were permeabilized and blocked in PBS containing 10% lamb serum and 0.1% triton x-100. Sections were immunostained with concomitantly expressed control mAb anti-DSG3 (Acc#: HQ338093.1 and HQ338094.1) (18µg/mL), MS39p2w174 (18µg/mL), or PBS in blocking buffer overnight at 4C. Sections were washed five times with PBS over 1 hour and counterstained with anti-human IgG for 1 hour at room temperature (Alexa Fluor 488, Jackson ImmunoResearch) (2µg/mL). Nuclei were stained with DAPI at 1:2000 for 5 minutes at room temperature. Rat oligodendrocyte precursor cells were prepared from rat embryos followed by panning and in-vitro differentiation into primary rat oligodendrocytes58. Cells on cover slips were permeabilized with ice-cold 100% methanol for 10min, blocked with 10% donkey serum for 1h at RT, and then stained with isotype control (anti-DSG3) or MS39p2w174 at 10µg/ml in 1% donkey serum for 1h at RT, before incubation with secondary Alexa Fluor 647 donkey anti-human IgG antibody (Jackson ImmunoResearch) for 1h at RT. Confocal images were taken with a Zeiss LSM 880 confocal microscope using ZEN software (Zeiss).
Bio-Layer Interferometry. Association and dissociation constants of mAbs to proteins and peptides were measured with bio-layer interferometry on an Octet QK device (Fortebio / Sartorius, Fremont, CA) according to standard protocols. For peptide kinetics, biotinylated peptides were bound to high precision streptavidin (SAX) biosensors (peptide concentration in solution: 100 nM) and mAbs MS39p2w174 and germline were probed as analytes in concentrations ranging from 10 – 270 nM. For protein kinetics, mAbs were bound to anti-hIgG Fc Capture (AHC) biosensors (mAb concentration in solution: 20 nM), and proteins were probed as analytes in concentrations ranging from 1.56 – 125 nM. Data was analyzed with BLI analysis software (Fortebio / Sartorius, version 7.1) and with GraphPad prism (version 8.4). Buffer controls were subtracted, and curves were fitted globally for each group consisting of all concentrations of the same ligand. KD values ± SD as well as association / dissociation curves were reported and plotted with GraphPad prism (version 8.4). KD, Kon, and Koff values are shown in Supplementary Table 6.
Prediction of Protein Disorder. Order and disorder along the amino acid sequences of EBNA1 and GlialCAM were analyzed with PONDR (Predictor of Natural Disordered Regions, WSU Research Foundation)59, using the VSL2 algorithm.
Crystallization of antibody-antigen complexes. EBNA1AA386-405 20mer peptides (>98% purity) (Sigma Aldrich) were mixed with MS39p2w174-Fab (15 mg/ml) in a 1:7.5 molar ratio and incubated overnight. Crystals for MS39p2w174-Fab + EBNA1AA386-405 grew in 0.48M Sodium Citrate, 0.72M Sodium/Potassium Phosphate, and 3% MPD (v/v) in 0.1M HEPES, pH 6.9 (Extended Data Fig. 6c). Crystals were harvested, cryo-protected with a quick dip in a cryo-solution containing the well solution with 25 % glycerol, and flash cooled in liquid nitrogen. Data were collected at beamline SSRL 12-2 at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, and processed and scaled using XDS/aimless and Staraniso60,61. Crystals belonged to space group I222 (a = 119.66 Å, b = 137.56 Å, c = 179.00 Å, α = β = γ = 90˚) and contained two Fab-peptide complexes per asymmetric unit (Extended Data Fig. 6d). Phaser was used for molecular replacement62 with the model structure 4LRI (PDB)63, stripped of all CDR loops. Loops were re-constructed with Coot 64 and structures were refined with phenix.refine 65,66 in iterations with Coot. Measurements and figure design was done with Pymol v2.1 67. The structure was deposited in the protein data bank (PDB, www.rcsb.org 26) with PDB ID: 7K7R.
Mouse Immunization, EAE, and histology. All animal experiments were performed in accordance with state and federal guidelines and regulations, and approved by the Stanford Institutional Animal Care and use Committee. 8-week-old female SJL/J and FVB x C57BL/6 mice were purchased from The Jackson Laboratory. The mice were housed in recyclable individually ventilated (IVC) cages, with a 12-light/hour dark cycle, at a temperature of 70 degrees F, and with 50% humidity. Mice were immunized s.c. with 200 µg/mouse of EBNA1AA386-405 (peptide sequence: SQSSSSGSPPRRPPPGRRPF) or scrambled control peptide (peptide sequence: SPSRPGRSRSRGSPFPQPSP) (10 mice per group), mixed with 100 µg/mouse of CpG (ODN1826, Invivogen, San Diego, CA) in 100 µl/mouse incomplete Freund’s adjuvant (BD Difco, Franklin Lakes, NJ). 3 weeks later EAE was induced by s.c. immunization with 200 µg/mouse of PLPAA139-151, mixed with the same peptides used in the first immunization, in 100µl of incomplete Freund’s adjuvant, supplemented with 200 µg/mouse of mycobacterium tuberculosis (strain H37 RA, BD Difco). Serum samples were obtained by retro-orbital blood draws 3 days before the 1st and 2nd immunizations (day -24 and day -3), and during termination of the experiment (day 50). Mice were weighed daily, and disease severity was assessed according to a 5-point standard scoring system: 0, no clinical signs; 1, loss of tail tone; 2, hind limb weakness; 3, complete hind limb paralysis; 4, hind limb and forelimb paralysis; 5, moribund or dead. Mice were euthanized on day 50 post induction of EAE by deep anesthesia with i.p. injections of 0.01 ml/g body weight 7.2% Xylazine (Bayer Healthcare, Leverkusen, Germany) and 10.8% Ketamine (Pfizer, New York City, NY). Lymph nodes and spleens were extracted, and mice were then perfused with 10 ml PBS and 20 ml 4% paraformaldehyde (PFA) (Electron Microscopy Sciences, Hatfield, PA). Brains and spinal cords were extracted, stored in 4% PFA overnight, followed by 30% sucrose in PBS. Tissues were embedded in paraffin, sectioned, and stained for H&E as well as Luxol fast blue according to standard protocols. Infiltration of inflammatory cells into the spinal cord on H&E slides was assessed by a blinded investigator by counting lesions of infiltrating cells per slide, taking lesion size into account: 1: small infiltrate (<10 cells), 2: medium infiltrate (<100 cells), 3: large infiltrate (>100 cells). Demyelination was assessed by a blinded investigator according to a histological score64: 0.5, single demyelinated spot; 1, several spots; 2, confluent sites of demyelination; 3, extensive demyelination, less than half of a spinal cord; 4, demyelination of more than half of the spinal cord; and 5, extensive demyelination affecting >85 % of the total white matter of the spinal cord.
In vitro human T cell stimulation with proteins and peptides. Cryopreserved PBMCs were thawed and stabilized overnight at 37°C. The cells were pre-incubated for 30 minutes at 37°C, 5% CO2 with Polymyxin B (Sigma Aldrich) at a concentration of 10 mg/mL. The cells were then incubated for 16 hours with 100 mM of each recombinant protein or peptide in the presence of 2 mg/ml of anti-human CD28 (clone CD28.2, BD Biosciences) and anti-CD49d (clone 9F10, BioLegend) antibodies and IL-2 (50 IU/ml, Peprotech, Cranbury, NJ) and IL-7 (5 ng/ml, Peprotech). To detect intracellular staining, eBioscienceTM Protein Transport Inhibitor Cocktail (500X, ThermoFisher Scientific) was added during the final 5 hours of culture. After 16 hours, the cells were labeled with Fixable Viability Stain 510 (BD Biosciences) for live cell staining and fluorophore conjugated anti-CD3 (clone SK7, BD Biosciences), CD4 (clone RPA-T4, BD Biosciences), CD8 (clone RPA-T8, BD Biosciences), Granzyme B (clone GB11, BD Biosciences), IFNg (clone B27, BD Biosciences) and IL-17A (clone BL168, BD Biosciences) antibodies and detected using a BD LSR Fortessa.
Data analysis and statistics. The publicly available dataset from Han et al.27 was searched for abundance of GlialCAM in MS lesions. The old accession number Q8N7I3 was found, which was annotated in 2008 as unknown hypothetical protein and has since been replaced by accession number Q14CZ8. GlialCAM was identified with 2.5 mean spectral counts (MSC) in control tissue, 1.3 MSC in chronic plaques, 1.8 MSC in acute plaques, and 8 MSC in chronic-active plaques.
GraphPad Prism version 8.4.1 and R version 3.6.1 were used for statistical analyses. Statistical tests used are indicated in the respective methods section or in the figure legends.
Supplementary References
44. Thompson, A. J. et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 17, 162–173 (2018).
45. Polman, C. H. et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 69, 292–302 (2011).
46. Wingerchuk, D. M. et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85, 177–189 (2015).
47. Tan, Y.-C. et al. High-throughput sequencing of natively paired antibody chains provides evidence for original antigenic sin shaping the antibody response to influenza vaccination. Clin. Immunol. 151, 55–65 (2014).
48. Blum, L. K. et al. Circulating plasmablasts are elevated and produce pathogenic anti-endothelial cell autoantibodies in idiopathic pulmonary arterial hypertension. Eur. J. Immunol. 48, 874–884 (2018).
49. Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
50. Alamyar, E., Duroux, P., Lefranc, M.-P. & Giudicelli, V. IMGT(®) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol. Biol. 882, 569–604 (2012).
51. Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
52. Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
53. Huerta-Cepas, J., Serra, F. & Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 33, 1635–1638 (2016).
54. Bern, M., Kil, Y. J. & Becker, C. Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinformatics Chapter 13, Unit13.20 (2012).
55. Robinson, W. H. et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat. Med. 8, 295–301 (2002).
56. Kuerten, S. Autoantibodies against central nervous system antigens in a subset of B cell–dominant multiple sclerosis patients. Proc. Natl. Acad. Sci 202011249, 2011249117 (2020).
57. Schubert, R. D. et al. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat. Med. 25, 1748–1752 (2019).
58. Emery, B. & Dugas, J. C. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb. Protoc. 2013, 854–868 (2013).
59. Obradovic, Z. et al. Predicting intrinsic disorder from amino acid sequence. Proteins 53 Suppl 6, 566–572 (2003).
60. Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
61. Tickle, I. STARANISO: use of a WebGL-based 3D interactive graphical display to represent and visualise data quality metrics for anisotropic macromolecular diffraction data. Acta Crystallogr. A Found. Adv. 75, e162–e162 (2019).
62. McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
63. Fouts, A. E. et al. Mechanism for neutralizing activity by the anti-CMV gH/gL monoclonal antibody MSL-109. Proc. Natl. Acad. Sci. U. S. A. 111, 8209–8214 (2014).
64. Lanz, T. V. et al. Tryptophan-2,3-Dioxygenase (TDO) deficiency is associated with subclinical neuroprotection in a mouse model of multiple sclerosis. Sci. Rep. 7, 41271 (2017).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}