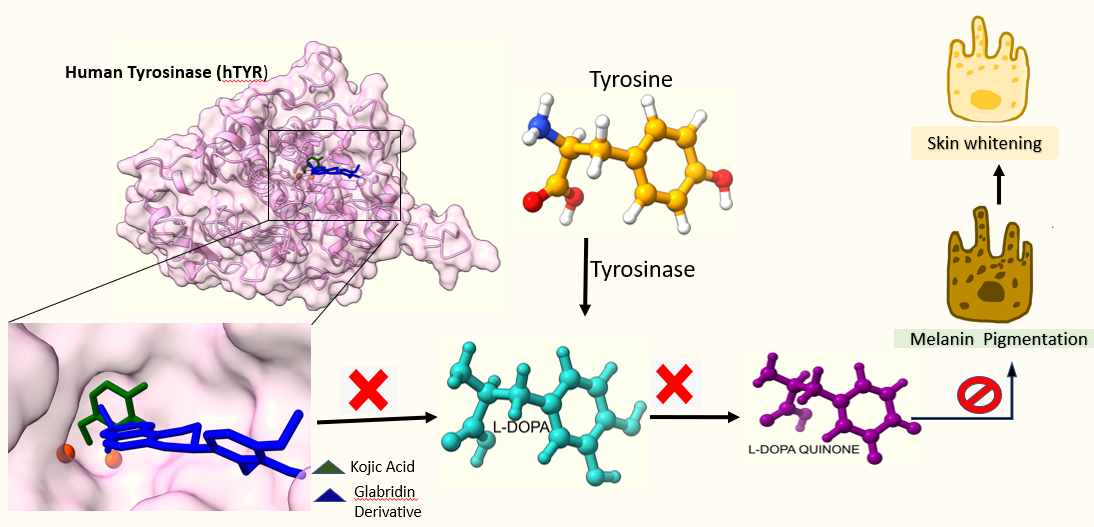
The effort was to find the anti-tyrosinase molecule using derivatives of glabridin. This study includes the natural compounds present in Glycyrhhiza glabra plant which are proposed to have anti-tyrosinase activity in traditional usages. All derivatives included in the study are (R)- glabridin [4484219], 5’prenyl glabridin [23641093], 2’,4’-O- dimethyl glabridin [480856], glabridin dimer [5481978], R- Hispaglabridin A [4484221], R-Hispaglabridin B [5318057], 5'-formyl glabridin [46230506], 4'-O-Methyl preglabridin [44257508], 4'-O-prenyl glabridin [480861], 4'-O-methyl glabridin [5319664], 3'-hydroxy-4'methoxy glabridin [10338211], 2'-O-methylglabridin [74830193], 2'-O,5'-C-diprenylglabridn [480868], Hispaglabridin A [ 442774] and Hispaglabridin B [15228661]. These are structural derivatives of glabridin and can be obtained from the extract of Glycyrhhiza glabra plant by HPLC purification except Glabridin dimethyl ether (2’,4’-O- dimethyl glabridin) and 2'-O-methylglabridin. These two compounds are semisynthetic and Belinky et al (1998) synthesized them by the following method. Glabridin (100 mg, 0.31 mmol) in a round bottom flask was dissolved in acetone (2 ml) and methyl iodide (30 ml, 0.33 mmol) and K2CO3 (64 mg, 0.46 mmol) were added. The reaction mixture was heated to 50°C with stirring, and after 6 h, the conversion reaction was determined by HPLC analysis. Glabridin (35%), 2’-O-methylglabridin (30%), 4’-O-methylglabridin (20%) and 2’,4’-O-dimethylglabridin (15%) were obtained. These compounds were separated and purified on a silica gel column, using CH2Cl2 and then CH2Cl2/3% CH3OH as eluents. 2’,4’-O-dimethylglabridin can be synthesized in a higher yield by using excess of methyl iodide and potassium carbonate in the initial reactions. Kojic acid was used as positive control for the docking studies and for in vitro studies glabridin (62% pure) was purchased from Kan Phytochemicals Pvt. Ltd.; Sonipat, Haryana. The experimental workflow employed is displayed in Fig 1.
Homology modelling of hTYR and secondary structure prediction
Due to non-availability of an experimentally established high – resolution X-ray crystal structure of hTYR in the Protein Data Bank, a three- dimensional structural model of the same was built using MODELLER (Webb et al, 2014) based on homology modelling. Tyrosinase protein sequence (Homo sapiens) (Accession no AAB60319.1) was retrieved from the NCBI protein database (https://www.ncbi.nlm.nih.gov) (Johnson et al, 2008) and used as query sequence to perform BLASTp. A structural model of the human tyrosinase was constructed using MODELLER using published crystal structure of 5, 6‐dihydroxyindole‐2‐carboxylic acid oxidase of human RCSB Protein Data Bank (Berman et al, 2007) (PDB ID: 5M8Q). The modelling template showed maximum 44.44 % identity and query coverage 81%. Modelling was done for the catalytic domain by removing 25 amino acids from the start along with a short sequence of 59 residues from 471 to 529 of the peptides. Two copper molecules were added to the catalytic centre obtained from Bacterial tyrosinase (Matoba et al, 2006). The overall quality of the modelled structure was validated through the Ramachadran plot using Rampage server. The secondary structure of the human tyrosinase enzyme sequence was also determined by self-optimized prediction method (SOPMA) (Combet et al, 2000). The secondary structure of protein is important to understand the three-dimensional structure of the protein and its function. The default parameters of window width 17, similarity threshold 8 and number of states 4 were used for this study.
Structure Preparation of Ligands and target hTYR
Glabridin and its derivatives (supplementary data-Table 1S) were retrieved from PubChem database to investigate their possible impact as inhibitor of human tyrosinase. 3D structures of glabridin derivatives were downloaded in sdf format. The sdf files were converted into pdbqt file format using open babel tool on Linux version 16.04.
Active site prediction and molecular Docking of ligands with Modelled hTYR
Active site prediction was performed by CastP web server (Tian et al, 2018). Docking studies were carried out on Autodock Vina (Trott and Olson, 2010) which carries out automated preparation steps for both receptor and ligands that include the addition of partial charges and required hydrogen atoms. Docking of ligand molecules was performed on predefined grid obtained from the template structure in complex with kojic acid (Lai et al, 2017). The grid size was 50 x 50 x 50 and dimension for X, Y and Z coordinates -15.715, 5.183 and -20.640, respectively. These parameters were also used to dock glabridin derivatives using Autodock Vina executed on Linux platform. The docking was validated by best possible conformation of each ligand and the best confirmation of each compound was selected based on the lowest docking score and their binding interactions measured in terms of Gibbs free energy (∆G). The docked structures (protein-ligand complexes) stabilize through several binding interactions such as polar, hydrophobic, hydrophilic, pi-pi stacking, salt bridge etc. The molecular interactions were analysed and evaluated based on their polar and hydrophobic interactions using structure visualization tool Pymol version 2.3 (DeLano, 2002), Chimera10.1 (Petterson, 2004) and LigPlot (Wallace et al, 1995).
Anti-melanogenic activity of glabridin on melanoma cell line
The anti-melanogenic activity of glabridin was evaluated on B16F10 cell line. The cytotoxicity of glabridin on murine melanoma cell lines (B16 melanoma cells) and effect of glabridin concentrations on the melanin production by melanoma cells was observed.
Effect of glabridin on Melanin production by B16 melanoma cells: Melanin contents in cultured B16 melanoma cells were measured according to the method of Oikawa et al. (1973) with slight modifications. B16 melanoma cells were seeded (initial density of cells 0.4 x 104 cells/cm2) in T75 culture flasks, and glabridin was added to the culture medium with concentrations from 1.0µg/mL to 5µg/ml. After 3-day culture, the cells were collected by brief trypsinization and subsequent centrifugation, and then treated with 5% trichloroacetic acid, ethyl alcohol: diethyl ether (3: 1) and diethyl ether successively in this order. The cells were dissolved with 2M NAOH containing 10% DMSO. Doxorubicin was used as positive control. The melanin contents were measured with Agilent Cary 50 UV Visible spectrophotometer at 400 nm. Results were analysed statistically using statistical software Prism 5 via one-way analysis of variance at P <0.05 with control.
Cell viability assay (MTT assay): B16F10 cells were grown to confluence in T75 cm2 flask supplemented with Dulbecco's Modified Eagle Medium and 10 % fetal calf serum in CO2 incubator with 5 % CO2. Cells were seeded at a density of 1x104 cells per well in DMEM medium. Twenty-four hours post seeding, cells were treated at concentrations ranging from 5µg/ml to 200µg/ml of glabridin for different time intervals (24 and 48 hr). After the exposure times, MTT was added to a final concentration of 0.5 mg/ml medium and the plates were incubated for 4 h at 37° C. The purple formazan crystals formed were dissolved in DMSO and read at 570 nm in a micro-quant plate reader. The assay was carried out in triplicates. The results were expressed as % inhibition.
Drug likeness and In-silico ADME prediction
Early detection of ADME properties reduces the failure in clinical phases and also minimises the load of synthesis of compounds for testing its potentiality for drug. Hence, it has become a vital tool in drug candidate identification. On this note, in silico prediction of the ADME properties (absorption, distribution, metabolism and excretion) was performed using SwissADME web tool (Daina et al, 2017) to determine the activity of these molecules within human body. These pharmacokinetic parameters were evaluated for the glabridin and its 15 derivatives to investigate their drug candidate chance. SMILES of each compound was obtained from PubChem database and was used for the analysis. The drug likeness of the molecules was predicted by adopting Lipinski’s Rule of 5. The rule of 5 predicts molecules with more than 5H-bond donors, 10-H bond acceptors, molecular weight more than 500 Da and the logP greater than 5 likely to had poor absorption and permeation of molecular entities (Ibrahim et al, 2020).
Molecular dynamics simulation study
MD simulation has been performed using GROMACS 5.1.2. (Berendsen et al, 1995) to analyse the structural stability of tyrosinase upon ligand binding, under GROMOS96 43a1 force field (Pol-Fachin et al, 2009). MD simulations for apo-protein (TYR) along with all of the receptor-ligand complexes, TYR-Kojic acid, TYR-5’- formylgrabridin, TYR-5’ prenylglabridin, TYR- Glabrdin dimer, TYR- 3’-hydroxy-4’-methoxyglabridin and TYR- (R)-glabridin, were performed in triclinic periodic boundary conditions. The topologies for ligand molecules were prepared using PRODRG (Schüttelkopf and Van Aalten, 2004). The systems were prepared with solvation using SPC water model at 1 nanometre marginal radius, followed by neutralization by adding the significant number of Na+ ions. The final systems, consisted of Protein, Ligand (except for TYR), Na+ ions and solvent, was subjected to energy minimization step for each system was performed using steepest decent integrator for 5000 iterations using the 0.01 energy step. After that, NVT (Berendsen et al, 1995) and NPT (Berendsen et al, 1984) ensembles were employed for temperature and pressure coupling and equilibration with leap-frag integrator for 50000 steps (100 ps). Finally, 50000 ps production simulations of each system were performed 2 fs time interval (Millan et al, 2017). The RMSD of backbone, RMSF of C-alpha atoms, SASA, Rg, and hydrogen bond were retrieved from MD simulation trajectories, and analysis plots were prepared using OriginPro.
{kind=link}