3.1. IFN-I signaling is required for CNS neuroinflammation following JEV inoculation in distal tissues
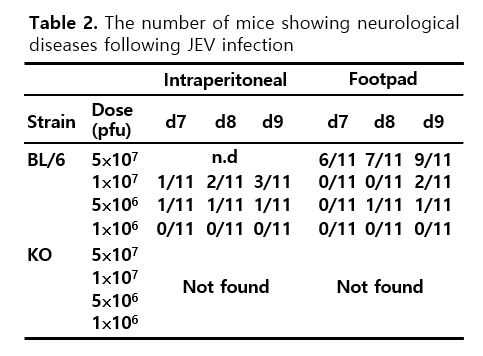
IFN-I signaling has been known to play a crucial role in conferring protection against various viral infections [20–22]. However, how IFN-I participates in viral dissemination and CNS neuroinflammation following JEV infection remains largely unknown. To test whether IFN-I signaling plays a role in inducing neurological disorders (JE) following JEV infection, IFN-I signal-competent (BL/6) and incompetent (IFNAR KO) mice were infected with JEV at different doses (1⋅106, 5⋅106, and 1⋅107 ffu) via the intraperitoneal or footpad route for systemic or local infection, respectively. As expected, IFN-I signal-incompetent mice show highly enhanced susceptibility to JEV infection, compared to IFN-I signal-competent mice (Fig. 1a). In contrast with IFN-I signal-competent mice, incompetent mice all succumbed to JEV infection within 6 and 7 days following JEV infection at a low dose (1⋅106 ffu/mouse) via the intraperitoneal and footpad route, respectively. Here, the interesting result was that IFN-I signal-competent mice infected with JEV (5⋅107 ffu/mouse) via the footpad route died through a prolonged and neurological illness that was symbolized by weight loss, distressed fur, and back hunching with postural imbalance, ataxia, and generalized tonic-clonic seizure, whereas IFN-I signal-incompetent mice all died without any neurological signs following JEV infection at different doses via the footpad route (Table 2). This result implies that while IFN-I signal-competent mice show typical JE signs of CNS neuroinflammation after JEV inoculation at the distal site, IFN-I signal-incompetent mice succumb to other fatal damages. To better understand fatal damages in IFN-I signal-incompetent mice following JEV infection, we determined the viral burden in various peripheral lymphoid and non-lymphoid tissues, as well as CNS tissues such as the brain and SC. IFN-I signal-incompetent mice were found to contain 102–104-fold increased levels of viral RNA in peripheral lymphoid and non-lymphoid tissues, including pLNs, spleen, mLNs, iliac LNs, BM, lung, liver, intestine, and kidney, 1, 2, and 3 days after JEV inoculation at footpad tissues, compared to IFN-I signal-competent mice (Fig. 1b). In contrast with peripheral lymphoid and non-lymphoid tissues, IFN-I signal-incompetent mice showed no significant increase in the levels of viral RNA in the SC and brain, even though they contained modestly increased viral loads in CNS tissues compared to competent mice (Fig. 1c). In addition, infectious JEV was recovered at higher levels from peripheral tissues such as the spleen and liver in IFN-I signal-incompetent mice than in the competent mice (Fig. 1d). However, infectious JEV was detected at levels with no significant increase in the brain of IFN-I signal-incompetent mice, compared to IFN-I signal-competent mice. These data indicate that IFN-I signaling plays a crucial role in restricting viral dissemination to peripheral tissues after the local inoculation of JEV. Conversely, our data also suggest that IFN-I signal is required to induce neurological disorders like JE as CNS neuroinflammation after JEV inoculation at distal sites such as the footpad.
3.2. Requirement of IFN-I signaling for CNS neuroinflammation through the restriction of JEV dissemination
To understand fatal damages and viral dissemination in IFN-I signal-incompetent mice following JEV infection, we performed several histopathological examinations and immunohistochemical studies for JEV Ags in various tissues including peripheral lymphoid and non-lymphoid tissues as well as CNS tissue such as the brain. Histopathological examinations revealed that IFN-I signal-incompetent mice had a destructive architecture in peripheral lymphoid tissues, such as pLNs, spleen, and mLNs, 2 days after the footpad inoculation of JEV (Fig. 2a). Notably, the spleen of IFN-I signal-incompetent mice showed reduced white pulp along with the depletion of immune cells, and the size of pLNs and mLNs was considerably reduced in IFN-I signal-incompetent mice. In addition, multifocal necrosis is observed in the whole liver of IFN-I signal-incompetent mice, along with dying cells accompanied by epithelial cells of the villi falling away in the intestine. In contrast with the peripheral lymphoid and non-lymphoid tissues, in both IFN-I signal-competent and incompetent mice, there was no apparent change in pathological phenomena in the CNS tissue 2 dpi, except that some hemorrhages were observed in the brain stem and adjacent parts of the cerebellum in IFN-I signal-incompetent mice. However, in histopathological examinations of IFN-I signal-competent mice at 6–7 dpi, we could find CNS neuroinflammatory reactions in brain tissues, such as some leukocyte infiltration (Fig. 2b). In addition, JEV Ags were more apparently detected in peripheral lymphoid and non-lymphoid tissues of IFN-I signal-incompetent mice compared to competent mice (Fig. 2c). We observed no significant JEV Ags in both the brains of IFN-I signal-competent and incompetent mice at 2 dpi (Fig. 2d). However, JEV Ags were detected in the brain of IFN-I signal-competent mice showing neurological disorders (Fig. 2e), which indicates that the JEV invasion of the CNS could be important in mice displaying neurological disorder. Taken together, these histopathological and immunohistochemical examinations support a crucial role of IFN-I signaling in eliciting CNS neuroinflammation by restricting viral dissemination to peripheral tissues following the local inoculation of JEV.
3.3. IFN-I-signaling restricts hemorrhage-like diseases following JEV infection
IFN-I signal-incompetent mice displayed viral dissemination in various peripheral organs and died without CNS neuroinflammation after distal JEV inoculation. Subsequently, we decided to further explore why IFN-I signal-incompetent mice succumbed so rapidly to JEV infection without CNS neuroinflammation. To this end, we analyzed their blood chemistries. The levels of red blood cells (RBC) and hemoglobin were found to significantly drop in IFN-I signal-incompetent mice at 24 and 36 h pi, however, white blood cell (WBC) levels were temporarily higher at an earlier time point of 6 h pi in IFN-I signal-incompetent mice compared with IFN-I signal-competent mice (Fig. 3a). In particular, the number of blood platelets in IFN-I signal-incompetent mice was persistently and markedly decreased from 24 h pi, which indicates that JEV infection caused thrombocytopenia in these mice (Fig. 3b). As a result, IFN-I signal-incompetent mice had lower hematocrit values at 24 and 36 h pi compared to competent mice. Because thrombocytopenia and hematocrit changes may be associated with the damage of major viscera organs [44, 45], we checked liver and kidney function. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, which are typical parameters for liver function, were at around 5-fold increased levels from 24 h pi in IFN-I signal-incompetent mice compared to competent mice (Fig. 3c). These increases in ALT and AST levels were very correlated with the occurrence of thrombocytopenia at 24 h pi. Similarly, the impaired parameters for kidney function were observed in IFN-I signal-incompetent mice, as shown by increases in blood urea nitrogen (BUN) and creatinine level in sera (Fig. 3d, left and middle graphs). The reduction of glucose level in sera of IFN-I signal-incompetent mice may also reflect impaired kidney function (Fig. 3d, right graph) because hypoglycemia is associated with renal failure [46], increase in glucose use [47, 48], and decreasing hepatic glucose production [47, 49]. The presence of thrombocytopenia and the damage of major viscera organs such as liver and kidney with fatal non-neurological disease suggested that the inoculation of JEV into the footpad might cause hemorrhage-like disease in IFN-I signal-incompetent mice [50–52]. Vascular leakage is a hallmark of hemorrhagic disease such as the severe form (dengue hemorrhagic fever, DHF) caused by dengue virus infection [50, 51]. Therefore, we checked whether the footpad inoculation of JEV could induce increased vascular permeability in IFN-I signal-incompetent mice. To assess the integrity of the vascular endothelial barrier, IFN-I signal-competent and incompetent mice were intravenously injected with Evans blue, a dye that preferentially binds to proteins such as albumin. Then, the extravasation of Evans blue dye into tissues was visualized in the intestine and liver following vigorous heart perfusion (Fig. 3e,f). As expected, IFN-I signal-incompetent mice showed increased extravasation of Evans blue dye, as shown by a greater intensity of dark blue in the intestine and liver. Notably, dispersed spots of Evans blue dye occurred in the entire liver tissues of IFN-I signal-incompetent mice, which indicates necrosis by JEV replication. Furthermore, we quantified the amount of Evan blue dye extravasated into liver and intestine tissues spectrophotometrically (Fig. 3g). Similarly, intestine and liver tissues of IFN-I signal-incompetent mice were observed to contain higher levels of extravasated Evan blue dye compared to competent mice. However, dissimilar to the results derived from intestine and liver tissues, brain tissue derived from IFN-I signal-competent and incompetent mice showed no significantly altered extravasation of Evan blue dye. Collectively, these results support the conclusion that impairment of IFN-I signaling by the distal inoculation of JEV, such as via the footpad route, drives hemorrhagic-like diseases rather than neurological diseases.
3.4. Altered expression of TJs and adhesion molecules results in increased vascular leakage in IFN-I signal-incompetent mice
The importance of TJs and adhesion molecules in restricting the invasion of non-neural tissues after JEV infection has been described [53]. Therefore, to further understand the increase of vascular leakage in the peripheral tissue of IFN-I signal-incompetent mice following peripheral JEV inoculation, we checked the expression of TJs and adhesion molecules. The mRNA levels of TJs including claudin-1, claudin-5, occludin, and ZO-1 was significantly reduced in liver and intestine of IFN-I signal-incompetent mice 2 days following footpad inoculation of JEV (Fig. 4a, b), but mRNA levels of TJs, except ZO-1, in brain tissue were almost the same in both IFN-I signal-competent and incompetent mice (Fig. 4c). The expression of the adhesion molecules (ICAM-1 and JAM) in the liver, intestine, and brain appeared complicated. ICAM-1 expression was observed to be higher in the liver and intestine of IFN-I signal-incompetent mice compared to competent mice. However, JAM expression was reduced in the liver of IFN-I signal-incompetent mice but increased in the intestine compared to competent mice (Fig. 4d). Also, ICAM-1 expression was increased in the brain of IFN-I signal-incompetent mice but appeared to be delayed compared to other organs. These results indicate that the expression of adhesion molecules could be differentially regulated in peripheral organs and the CNS, depending on the progression of the disease. Conclusively, the reduction in the TJ expression in the liver and intestine tissues of IFN-I signal-incompetent mice supports an increase of vascular leakage in peripheral organs, and the altered expression of adhesion molecules in peripheral organs and brain may affect infiltration of leukocytes in inflamed tissues.
3.5. IFN-I signaling restrains the “cytokine storm” following JEV infection
To further understand the basis of peripheral organ damage, such as that in the liver and kidney, in IFN-I signal-incompetent mice after the footpad inoculation of JEV, we measured the expression of several pro-inflammatory cytokines and chemokines in various peripheral tissues including pLN, spleen, mLN, iLN, and liver, as well as in CNS tissues including SC and brain. As a result, IFN-I signal-incompetent mice showed persistent and highly increased levels of TNF-α, IL-6, CCL2, and CXCL2 mRNA expression in the various tissues tested at the periphery from 24 h pi, compared to IFN-I signal-competent mice (Fig. 5a). Similarly, these cytokines (TNF-α, IL-6) and chemokines (CCL2, CXCL2) were expressed in the CNS tissues of IFN-I signal-incompetent mice with higher levels than those of competent mice, even though the expression of pro-inflammatory cytokines in the CNS tissues were somewhat low and delayed compared to other peripheral tissues (Fig. 5b). In addition, we measured serum levels of vasoactive IL-6 and TNF-α proteins, which have been known to play an important role in inducing hemorrhagic fever and increasing vascular leakage [51, 54]. As expected, the levels of serum IL-6 and TNF-α were increased by 5- to 60-fold from 24 h pi, in IFN-I signal-incompetent mice compared to competent mice (Fig. 5c). The massive increase of multiple CC chemokines in sera of IFN-I signal-incompetent mice was also revealed, when we checked the levels of multiple CC chemokines in sera with CBA methods (Fig. 5d). This massive and uncontrolled production of multiple cytokines and chemokines in IFN-I signal-incompetent mice after JEV footpad inoculation was coupled with renal and hepatic injury and JEV Ags staining in tissues, and suggests a picture of sepsis due to “cytokine storm”.
3.6. IFN-I signal is essentially required for quick innate IFN-I responses
To better understand IFN-I innate responses in IFN-I signal-incompetent mice, we measured the expression of IFN-I (IFN-α/β) mRNA in various peripheral tissues including pLN, spleen, mLN, iLN, and liver after JEV footpad inoculation. Our data revealed that the expression of IFN-α/β mRNA in the peripheral tissues of IFN-I signal-incompetent mice increased 5 to 500 times at 48 and 72 h pi compared to the IFN-I signal-competent mice (Fig. 6a). Similarly, the CNS tissues (SC and brain) in IFN-I signal-incompetent mice displayed a higher expression of IFN-α/β mRNA 72 h after JEV footpad inoculation than IFN-I signal-competent mice (Fig. 6b). This implies that the ablation of the IFN-I signal did not induce blunted IFN-I production after JEV footpad inoculation and, rather, the magnitude of IFN-I (IFN-α/β) expression seemed to follow the degree of JEV replication because the replication of JEV was coupled to the expression levels of IFN-α/β. IFN-I (IFN-α/β) binds to a heterodimeric receptor (IFNAR) and mediates downstream pleiotropic functions of the canonical JAK-STAT signaling pathway [23, 24]. This stimulation results in the induction of antiviral ISGs, induction of cell surface and cytosolic PRRs in antigen-presenting cells, and the regulation of cytokine and chemokine production [19]. Therefore, we measured the induction levels of antiviral ISGs to define IFN-I innate responses in greater detail. We specifically focused on the induction of PRRs (RIG-I [DDX1], MDA5 [IFITH1]), their transcription factors (IRF3, IRF7), and ISGs (ISG49 [IFIT3], ISG54 [IFIT2], ISG56 [IFIT1]). As a result, the expression of PRRs (RIG-I, MDA-5) was markedly elevated in IFN-I signal-competent mice compared to incompetent mice (Fig. 6c). Notably, RIG-I and MDA5 were induced by approximately 70-fold increased levels in the liver of IFN-I signal-competent mice at 3 dpi. Subsequently, IRF3 and IRF7 expression followed expression patterns of PRRs (RIG-I, MDA-5) in IFN-I signal-incompetent mice, except for IRF3 expression in brain tissue (Fig. 6d). The expression of ISGs (ISG49, ISG54, ISG56) was observed and peaked quickly at 1 dpi in IFN-I signal-competent mice, but IFN-I signal-incompetent mice showed no induction of ISG expression (Fig. 6e). Collectively, these results suggest that the impaired induction of ISG expression in the peripheral tissues, such as the liver, of IFN-I signal-incompetent mice at a very early stage was likely to contribute to the appearance of a hemorrhagic-like disease, rather than CNS encephalitis.
3.7. Intrinsic IFN-I signaling in tissue-resident cells is critical to limiting hemorrhage-like diseases following JEV infection
IFN-I signal-incompetent mice displayed hemorrhage-like disease rather than CNS encephalitis after JEV inoculation at the distal site, and this altered disease appeared to be caused by impaired innate responses at a very early stage. However, the cell types that mainly contribute to the altered disease in IFN-I signal-incompetent mice remain to be defined. Therefore, we were interested in dissecting the contribution of tissue-resident cells and HSC-derived leukocytes in this process. To achieve this, we used a BM chimeric model established using IFN-I signal-competent (BL/6) and incompetent (IFNAR KO) mice. Strikingly contrasting results were revealed in our BM chimeric experiments. IFNAR KO recipients of BL/6 WT BM cells (BL/6–KO chimera) or IFNAR KO BM cells (KO–KO chimera) showed the same susceptibility to JEV footpad inoculation, whereas BL/6 WT recipients of IFNAR KO BM cells (KO–BL/6 chimera) or BL/6 WT BM cells (BL/6–BL/6 chimera) displayed almost the same resistance to JEV infection (Fig. 7a left graph). Furthermore, the BL/6–KO chimera died without neurological disorders like the KO–KO chimera, but the KO–BL/6 chimera succumbed to neurological disorders that showed higher frequencies than the BL/6–BL/6 chimera (Fig. 7a, right graph). To better understand this contrasting result in the BM chimeric experiments, we checked the viral load in various peripheral tissues including pLN, spleen, liver, and intestine, as well as in CNS tissues such as the brain. In support, similar to KO–KO chimera, BL/6–KO chimera showed markedly increased loads of JEV in various peripheral tissues 48 h pi, as compared to both KO–BL/6 and BL/6–BL/6 chimeras (Fig. 7b). No significant differences in JEV loads in peripheral tissues were observed between KO–KO and BL/6–KO chimeras. However, both BL/6–KO and KO–KO chimeras showed no significantly increased loads of JEV in CNS tissues, such as the brain, compared to those of KO–BL/6 and BL/6–BL/6 chimeras, which indicates that tissue-resident cells play an important role in disseminating JEV into the peripheral lymphoid and non-lymphoid tissues after JEV footpad inoculation.
Next, we checked the expression of pro-inflammatory cytokines and chemokines in the peripheral lymphoid and non-lymphoid tissues as well as in CNS tissues, to further characterize the disease induced in BM chimeric models after JEV footpad inoculation. BL/6–KO chimera showed highly enhanced levels of pro-inflammatory cytokines (IL-6, TNF-α) and chemokines (CCL2, CCL3, CCL4, CXCL2) in the peripheral tissues, as shown in KO–KO chimera (Fig. 7c). However, the expression of pro-inflammatory cytokines and chemokines in the brain was at almost the same low level in the four BM chimeric models, except for certain chemokines such as CCL2, CCL3, and CCL4. Similarly, pro-inflammatory cytokine and chemokine proteins in the sera of the BL/6–KO chimera were detected at markedly higher levels compared to KO–BL/6 and BL/6–BL/6 chimeras (Fig. 7d), which indicates that the BL/6–KO chimera displayed a “cytokine storm” after JEV footpad inoculation, as occurred in IFN-I signal-incompetent mice. Because the IFN-I signal-incompetent mice showed hemorrhage-like disease through thrombocytopenia and altered hematocrit after JEV footpad inoculation, we checked blood chemistries of the four BM chimeric models. The marked reductions in the values of RBC, hemoglobin, platelet, and hematocrit were observed in both BL/6–KO and KO–KO BM chimeras, as compared to BL/6–BL/6 and KO–BL/6 chimeras (Fig. 7e). Notably, BL/6–KO BM chimera have severe thrombocytopenia, thereby resulting in a reduction of hematocrit value, as shown in IFN-I signal-incompetent mice after JEV footpad inoculation. Furthermore, to evaluate whether the BL/6–KO BM chimera had the peripheral organ injury found in IFN-I signal-incompetent mice, we checked liver and kidney function in the four BM chimeras after JEV footpad inoculation. As expected, the BL/6 KO BM chimera had highly enhanced values of ALT and AST, which indicates that the BL/6–KO BM chimera has impaired liver function after JEV infection (Fig. 7f). Similarly, increased creatinine and BUN values in the BL/6–KO BM chimera indicated impaired kidney functions and displayed hypoglycemia, as shown in IFN-I signal-incompetent mice after JEV infection. Finally, we assessed the integrity of the vascular endothelial barrier using the Evan blue dye extravasation method. Enhanced vascular leakages confirmed that the BL/6–KO BM chimera had the hemorrhage-like disease shown in IFN-I signal-incompetent mice after JEV footpad inoculation (Fig. 7g). Summarizing all the results derived from the BM chimeric experiments, our data suggest that IFN-I signaling in tissue-resident cells plays a critical role in restricting hemorrhage-like disease after JEV inoculation at the distal site.
3.8. IFN-I signaling attenuates JEV replication in hepatocytes, enterocytes, and neuron cells
The intrinsic role of IFN-I signal in tissue-resident cells, rather than HSC-derived cells, in restricting viral dissemination and hemorrhage-like disease after JEV footpad inoculation was proposed. Therefore, we were interested in the effect of IFN-I signaling on JEV replication in tissue-resident cells, in order to define cellular factors for viral dissemination and hemorrhage-like disease in IFN-I signal-incompetent mice. To achieve this, we prepared primary hepatocytes, enterocytes, and cortical neuron cells from IFN-I signal-competent and incompetent mice, and then infected those primary cells to check viral permissiveness. JEV was observed to replicate 10 to 100-fold more rapidly in primary hepatocytes, enterocytes, and neuron cells derived from IFN-I signal-incompetent mice compared to IFN-I signal-competent mice (Fig. 8a). Notably, JEV replicated much more quickly at 105 to 108-fold increased levels in primary neuron cells, compared to other primary cells including hepatocytes and enterocytes. This result indicates that IFN-I signaling is very important in regulating JEV replication in tissue-resident cells including hepatocytes, enterocytes, and neuron cells, and demonstrates that primary neuron cells are inherently and very permissive to JEV replication, compared to other tissue-resident cells such as hepatocytes and enterocytes. In support, infectious JEV was recovered with highly increased levels in primary hepatocytes, enterocytes, and neuron cells derived from IFN-I signal-incompetent mice compared to competent mice, and primary neuron cells produced infectious JEV with higher levels than other primary cells (Fig. 8b). To better understand the rapid replication of JEV in tissue-resident cells derived from IFN-I signal-incompetent mice, we examined the induction of antiviral ISGs in primary hepatocytes and neuron cells after JEV infection. As expected, primary hepatocytes derived from IFN-I signal-incompetent mice showed highly impaired induction of antiviral ISGs including MDA5, RIG-I, IRF3, and IRF7, as well as ISG49, ISG54, and ISG56, depending on the infectious dose of JEV (Fig. 8c). Primary neuron cells derived from IFN-I signal-incompetent mice also showed reduced induction of antiviral ISGs following JEV infection (Fig. 8d). Collectively, these results suggest that tissue-resident cells derived from IFN-I signal-incompetent mice are very permissive to JEV replication, due to impaired induction of antiviral ISGs. In addition, our data imply that neuron cells are inherently and highly vulnerable to JEV infection compared to other tissue-resident cells.
3.9. Early and higher infection of circulating CD11b + Ly-6C + monocytes at the inoculation site is a prerequisite to JEV dissemination
Tissue-resident cells that are deficient in IFN-I signaling showed high permissiveness to JEV infection and played a dominant role in inducing hemorrhage-like disease in IFN-I signal-incompetent mice after JEV footpad inoculation, due to enhanced JEV dissemination into peripheral organ tissues. Here, we were interested in identifying cellular factors that carry the virus throughout the body after JEV inoculation at the distal peripheral sites. To this end, we screened various cell types that could be infected in footpad tissue at an early phase (24 and 48 h pi) after JEV inoculation. Eventually, we found that CD11b+ myeloid cells were preferentially infected 48 h after JEV infection in the footpad tissues of IFN-I signal-incompetent mice compared to competent mice (Fig. 9a). Furthermore, our analysis revealed that CD11b+Ly-6C+ monocytes were highly infected in footpad CD11b+ myeloid cell population derived from IFN-I signal-incompetent mice. Notably, CD11b+Ly-6C+ inflammatory macrophages that show mature macrophage phenotype (F4/80-positive) in the footpad of IFN-I signal-incompetent mice were infected with highly increased levels, as shown by the high frequency of CD11b+Ly-6C+F4/80+ inflammatory macrophages with an NS1 JEV Ag-positive response. Similarly, the footpad derived from IFN-I signal-incompetent mice contained highly increased levels of NS1-positive CD11b+Ly-6C+F4/80+ inflammatory macrophages at 24 h pi compared to competent mice (Fig. 9b). Subsequently, we examined JEV-infected CD11b+Ly-6C+ monocytes in peripheral organ tissues such as the liver after JEV inoculation at the distal footpad tissue. CD11b+Ly-6C+ monocytes derived from the liver of IFN-I signal-incompetent mice were infected at higher levels compared to competent mice (Fig. 9c). However, unlike footpad tissue, liver CD11b+Ly-6C+F4/80+ inflammatory macrophages matured from monocytes had a comparable infection rate in both IFN-I signal-competent and incompetent mice. In support, JEV-infected CD11b+Ly-6C+ monocytes were detected at higher levels in the liver of IFN-I signal-incompetent mice compared to competent mice 24 h after JEV footpad inoculation, while CD11b-negative non-myeloid cells showing NS1 JEV Ag-positive response were detected at comparable levels (Fig. 9d). The next day (48 h pi), liver tissue derived from IFN-I signal-incompetent mice contained at high levels of JEV-infected CD11b+Ly-6C+ monocytes as well as CD11b-negative non-myeloid cells compared to competent mice. These results indicate that CD11b+Ly-6C+ monocytes and their matured macrophages could be preferentially infected in footpad tissue at an early stage in JEV infection, after which JEV-infected monocytes appear to carry the virus to peripheral organs, such as the liver. CD11b+Ly-6C+ monocytes that are recruited into inflamed tissues such as the footpad differentiate into macrophages, and some monocytes escape the inflamed tissues and enter the blood, thereby spreading the virus to entire peripheral organs [55–57]. In support of this, when we analyzed JEV-infected CD11b+Ly-6C+ monocytes in the blood, our results revealed that blood CD11b+Ly-6C+ monocytes were infected with highly increased levels in IFN-I signal-incompetent mice compared to competent mice 24 h after JEV footpad inoculation (Fig. 9e).
To better understand the preferential infection of JEV in CD11b+Ly-6C+ monocytes, we measured JEV loads in CD11b+Ly-6C+ monocytes purified from footpad, blood, and liver. As expected, CD11b+Ly-6C+ monocytes purified from the footpad, blood, and liver of IFN-I signal-incompetent mice contained 2 to 3-fold increased levels of JEV RNA compared to IFN-I signal-competent mice (Fig. 10a). In support, CD11b+Ly-6C+ monocytes purified from footpad, blood, and the liver of IFN-I signal-incompetent mice showed impaired induction of antiviral ISGs (MDA5, RIG-I, IRF3, IRF7, ISG49, ISG54, ISG56) compared to IFN-I signal-competent mice (Fig. 10b). Finally, we purified CD11b+Ly-6C+ monocytes from the blood of IFN-I signal-competent and incompetent mice to examine infectious JEV production in CD11b+Ly-6C+ monocytes. Our data revealed that CD11b+Ly-6C+ monocytes purified from IFN-I signal-incompetent mice produced higher levels of infectious JEV than IFN-I signal-competent mice (Fig. 10c). Taken together, these results suggest that preferential infection of CD11b+Ly-6C+ monocytes in the footpad tissue of IFN-I signal-incompetent mice is required for viral dissemination into the entire body. At an early stage after JEV footpad inoculation, JEV-infected CD11b+Ly-6C+ monocytes in the footpad are likely to carry the virus through the blood into peripheral tissues.
{kind=link}