Detailed descriptions of the methods can be found in Additional file3.pdf.
Cell culture
MDA-MB-231 (HTB-26), BT-549 (HTB-122), HCC-1806 (CRL-2335) and HEK293FT (PTA-5077) were obtained from ATCC (LGC Standard GmbH, Wesel, Germany). MDA-MB-231 and HCC-1806 were cultured in RPMI1640 medium supplemented with 10% FBS and 1% L-Glu. BT-549 was maintained in RPMI1640 medium supplemented with 10% FBS, 1% L-Glu and 0.1% Insulin. HEK293FT was cultured in DMEM supplemented with 10% FBS, 1% L-Glu, 1% NEAA and 1% Geneticin.
All parental cell lines were incubated at 37°C with 5% CO2, tested for potential mycoplasma contamination on a regular basis, and were positively authenticated prior to and in the end of the study (Multiplexion GmbH, Heidelberg, Germany).
Transfection
Cells were seeded at 70-80% confluency one day before transfection. All transfections (plasmids, siRNAs or miRNA mimics) were performed using Lipofectamine2000 (LF2000) according to the manufacturer’s instructions (0.4 µL/well in 96-well plates, 4 µL/well in 6well plates and 10 µL/dish in 10 cm dishes). Cells were incubated for 48 or 72 h at 37°C, 5% CO2 in a humidified atmosphere before they were used for experiments.
All siRNAs (Dharmacon, Lafayette, USA) and synthesized miRNA mimics (Qiagen, Hilden, Germany) were used at a final concentration of 30 nM. The siRNAs purchased from siTOOLs Biotech (Planegg, Germany) were used at a final concentration of 2 nM. The sequences of siRNAs and miRNA mimics are listed in Additional file 2: Supplementary Table 1.
Generation of stable cell lines
To generate TNBC cell lines overexpressing pre-miRNAs, the pre-miRNAs were cloned into a retroviral vector RT3GEPIR [19] and retroviral particles were produced in HEK293FT cells by co-transfection of the retroviral RT3GEPIR vector, together with the VSV.G envelope plasmid pMD2.G (Addgene #12259) and gag/pol packaging plasmid pHIT60 [20].
To generate inducible E2F1 overexpressing TNBC cell lines, the open reading frame of human E2F1 (Addgene plasmid # 70329) was shuttled into the lentiviral expression vector rwSMART-TRE3G-GW-mCMV-TetON3G (Cellular Tools DKFZ, Heidelberg, Germany) using Gateway cloning technology [21] (ThermoFisher, Braunschweig, Germany). Lentiviral particles were produced in HEK293FT cells by co-transfection of the lentiviral rwSMART-TRE3G-E2F1-mCMV-TetON3G expression vector, together with 2nd generation viral packaging plasmids VSV.G (Addgene #14888) and psPAX2 (Addgene #12260).
Virus-containing supernatant was collected 24 h after transfection. Centrifuged and filtered supernatant was used to transduce target cells in the presence of 10 µg/mL polybrene (Merck, Germany). 24 h after transduction, virus-containing medium was replaced with full growth medium containing 2 µg/mL puromycin.
Xenograft assay
3x106 MDA-MB-231 cells in 30 µL PBS:Matrigel (Corning, Bedford, USA, growth factor reduced, 1:1, v/v) stably overexpressing pre-miRNA (pre-miR-183 or two different pre-miRNA negative controls) were injected under isoflurane anesthesia into the 3rd mammary gland fat pad of 6-7 week-old female NSG mice (n=6/group). One week after inoculation, doxycycline (1 mg/mL in drinking water supplemented with 5% saccharose) was given. The Kliba 3307 and the drinking water including ingredients were replaced once every week throughout the study. Twice a week, the tumor size was measured by caliper in two dimensions. The weight of mice was recorded once weekly. Mice were followed up for 12 weeks and sacrificed once the tumor reached 1 cm in one diameter or if an alternative predefined humane endpoint was reached. Lungs were collected and checked for micrometastasis by Alu PCR [22]. The animal experiment was licensed under G288/14 by local regulatory authorities.
Transwell-based cell migration and invasion assay
Cells were starved in 0% FBS starvation medium for 24 h. Then, 100,000 cells in 200 µL of 0% FBS starvation medium were seeded into the upper compartment of Transwell inserts (Corning, Kaiserslautern, Germany). Full growth medium was used in the lower compartment as chemoattractant. Cells were allowed to migrate or invade for 16 h. In parallel, a black clear-bottom 96-well plate was prepared as a seeding control plate for normalization. The cells on the lower side of the membrane were fixed with 4% PFA for 15 min. Migrated cells and seeding control plate were stained with Hoechst 33342 and imaged with a Molecular Devices Microscope IXM XLS (Molecular Device, California, USA) using 4x S Fluor objective. Nuclei were defined by Hoechst signals within a certain size (6-35 µm) and intensity (5000 gray levels above local background) and counted using Molecular Devices Software (Molecular Device, California, USA). Afterwards, the exemplary membranes were stained with 0.5% crystal violet for 30 min and then imaged with a light microscope.
Cell viability assay
Cell viability was analyzed with a microscope-based nuclei counting method. Briefly, cells were seeded into black clear-bottom 96-well plates and transfected 24 h after seeding using Lipofectamine 2000. At different time points, cell nuclei were stained with Hoechst 33342 for 30 min and propidium iodide (20 ng/well, Thermo Fischer Scientific, Massachusetts, USA) for 15 min. Subsequently, the plates were imaged with Molecular Devices Microscope IXM XLS (Molecular Devices, California, USA) using 4x S Fluor objective. The cell number was obtained by counting cell nuclei on each image. Nuclei were defined by Hoechst signals within a certain size (6-35 µm) and intensity (5000 gray levels above local background), counted and automatically classified for positivity in the propidium iodide channel with Molecular Devices Software (Molecular Device, California, USA). The mean value of six technical replicates was used for each biological replicate.
BrdU/7ADD-based cell cycle assay
Cell cycle phases were analyzed with Bromodeoxyuridine (BrdU) and 7-Aminoactinomycin D (7-AAD) according to the manufacturer’s instructions. Briefly, cells were starved in 0% FBS starvation medium for 24 h, released from cell cycle block with full growth medium for 24 h and incubated with 10 µM BrdU 2 h prior to harvest. Cells were permeabilized by the Perm/Wash buffer and fixed with 250 µL Cytofix/Cytoperm buffer for 20 min at room temperature and incubated with 300µg/mL DNase for 1 h at 37°C. Cells were stained with Anti-BrdU antibody and 7-AAD and analyzed using a FACSCalibur device and CellQuest Pro (BD Biosciences, USA) and BD FACS DIVA software (BD Biosciences, USA).
RNA extraction and qRT-PCR
RNA was isolated using the RNeasy or miRNeasy Kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations. The concentration of total RNA was determined by NanoDrop ND-1000.
cDNA for mRNA analysis was prepared using the RevertAidTM H minus First-strand Kit (Thermo Fischer Scientific, Massachusetts, USA). Primers and probes are listed in Additional file 2: Supplementary Table 3. For quantification of miRNAs, miScript RT and PCR system (Qiagen, Hilden, Germany) was used. Raw data analysis was performed by using QuantStudio PCR Systems (Applied Biosystems). Data acquisition and raw data analysis were performed using QuantStudio PCR Systems (Applied Biosystems) with the ΔΔCt method [23].
Protein isolation and Western blotting
Cells were seeded in 6-well plates for pre-treatment (miRNA mimics and siRNAs). After treatment, the cells were lysed with RIPA lysis buffer (Thermo Fisher Scientific, Massachusetts, USA) containing Complete Mini protease inhibitor cocktail and PhosSTOP phosphatase inhibitor (Roche Applied Science, Penzberg, Germany). The protein concentrations of the samples were determined by the BCA Protein Assays Kit (Thermo Fisher Scientific, Massachusetts, USA) and quantified with a GloMax microplate reader (Promega GmbH, Walldorf, Germany).
The primary and secondary antibodies used in this study are listed in Additional file 2: Supplementary Table 4. The membranes were scanned and probed using the Odyssey Infrared Imaging System (LI-COR Biosciences, Nebraska, USA). The signal intensity of the band was quantified by using ImageStudio software and median background subtraction (LI-COR Biosciences, Nebraska, USA).
Mass spectrometry
Protein samples (10 µg per sample) were submitted to the DKFZ Genomics and Proteomics Core Facility for mass spectrometry-based protein analysis. Briefly, unfractionated samples were used for in-gel digestion on a DigestPro MSi robotic system (INTAVIS Bioanalytical Instruments) [24]. Peptides were separated on a cartridge trap column and eluting peptides were analyzed online by a coupled Q-Exactive-HF-X mass spectrometer (Thermo Fisher Scientific, Massachusetts, USA) running in the data depend acquisition mode.
Raw data was analyzed by the MaxQuant computational platform (version 1.6.3.3) using an organism-specific database extracted from Uniprot.org under default settings. Quantification was done by using a label-free quantification (LFQ) approach based on the MaxLFQ algorithm [25]. The Perseus software package (version 1.6.13.0) was used for imputation of missing values at default settings and statistical analysis [26].
Luciferase reporter assay
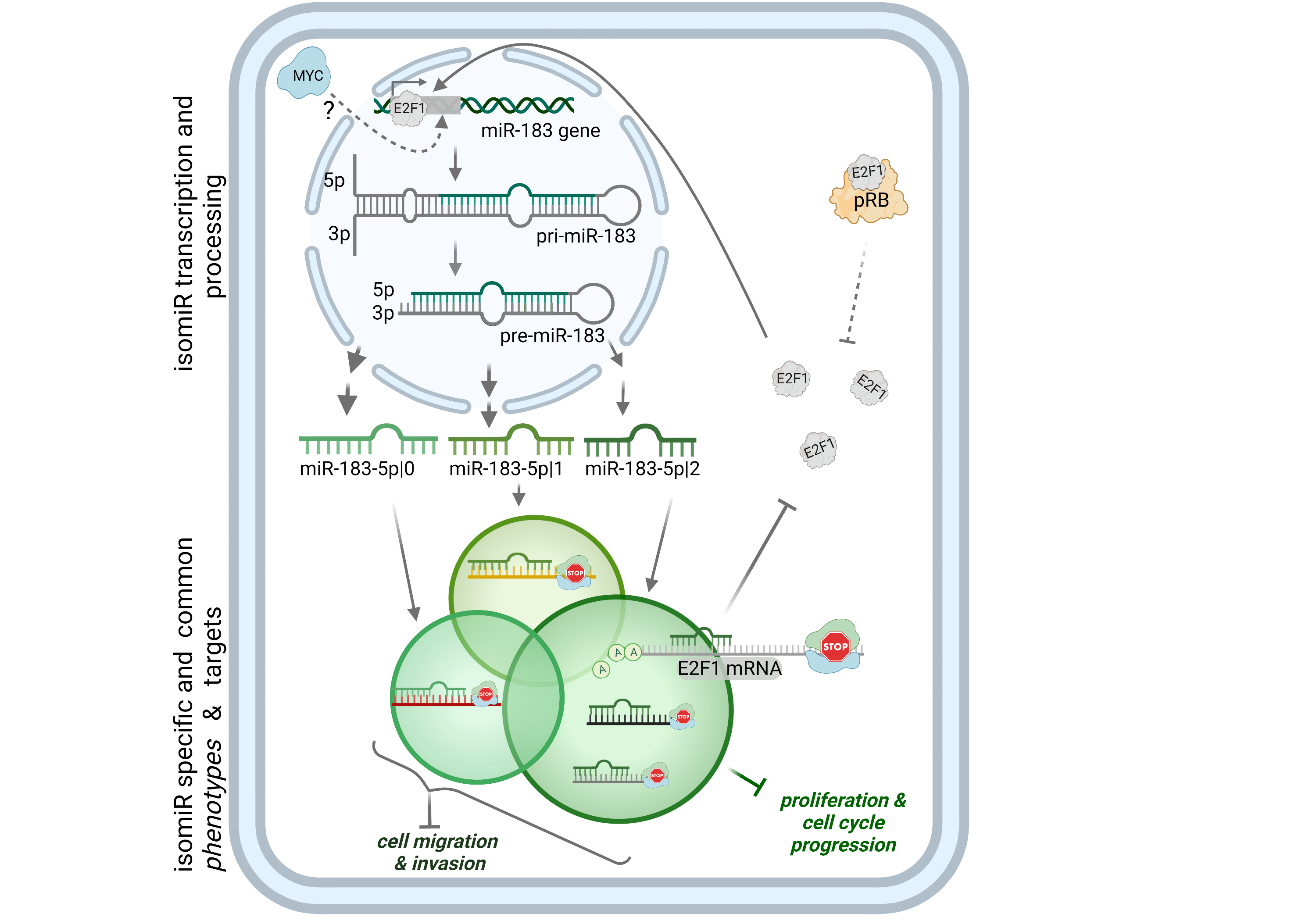
Direct targets of a miRNA of interest were validated by a 3’UTR dual luciferase reporter assay. The 3’UTR of E2F1 was cloned into psiCHECK-2 and subjected to site-directed mutagenesis of the predicted seed match for miR-183-5p|+2. The sequences of primers for cloning are listed in Additional file 2: Supplementary Table 2.
1.2x104 cells were seeded in white 96-well plates and transfected with miRNA mimics and 3’UTR reporter plasmid. 48 h after transfection, cells were washed and lysed and a dual luciferase assay was performed using a GloMax Microplate Reader (Promega Gmbh, Walldorf, Germany). The compositions of the buffers used in the luciferase assay are listed in Additional file 2: Supplementary Table 5.
miRNA target prediction
The 3’UTR sequences for the expressed genes were extracted using GEO dataset GSE27003 [27]. Reads from cell lines BT-20, BT-474, MCF7, MDA-MB-231, MDA-MB-468, T-47D, ZR-75-1 were aligned using the STAR (version 2.7.3a) algorithm [28] to the hg38 genome assembly and gencode v22 gene model. Reads aligning to 3’UTR regions of a gene were merged to get the expressed 3’UTR sequence. Non-overlapping regions found in the same UTR definition as provided by the gencode models were considered separately.
MiR-183-5p and its 5’isomiRs were subjected to target prediction by using miRanda (version 3.3a) [29] and Targetscan (version 7.1) [30] algorithms to the expressed 3’UTR sequences. A consensus set of miRNA-targets, i.e. an overlap of transcript/miRNA pair between the two prediction algorithms, was computed based on the principle of complementing algorithms as described by Riffo-Campos et al [31]. Venn diagrams visualizing the overlap between the target predictions for different isomiRs were created using an online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
TCGA and METABRIC patient data analysis
Breast Cancer (BRCA) expression quantification data for mRNA and miRNA were obtained from The Cancer Genome Atlas (TCGA) Research Network (https://www.cancer.gov/tcga). mRNA gene expression data (FPKM-UQ) were downloaded from the Genomic Data Commons (GDC) harmonized database (https://portal.gdc.cancer.gov/projects/TCGA-BRCA) using the Bioconductor R package TCGAbiolinks (version 2.12.6) and the GRCh38 build (hg38). Batch corrected isomiR expression data was obtained from GEO dataset GSE164767 [32]. All isomiR reads with an identical 5’ position resulting in an identical seed sequence were collapsed, i.e. summed up, to only further investigate 5’isomiR variants and isomiRs with a median expression of >15 reads per million (RPM) were considered for analysis. Only data from patients with corresponding mRNA expression and miRNA expression data were considered for further analysis. TNBC classifications were derived from Lehmann et al. and Koboldt et al. ([5, 33]). Patients classified as negative for ER, PR and HER2 receptors in the study by Koboldt et al. were labelled as TNBC patients. Patients lacking information for one of the three receptors were labelled ‘equivocal’. In case of clear labelling as TNBC in the Lehmann et al. study; ‘equivocal’ patents were re-labelled as TNBC. mRNA and miRNA expression data as well as patient information containing TNBC status from the METABRIC study were downloaded from https://ega-archive.org/dacs/EGAC00001000484/ and https://ega-archive.org/studies/EGAS00000000122 [34, 35].
To compare differences in isomiR expression in levels in the TCGA data between tumor and normal tissue, average expression in tumor tissue was divided by expression in matched non-tumor/normal tissue for all patients and the results were log2 transformed and the p-adjusted values were computed from an unpaired, two sample t-test (adjustment by the Benjamini-Hochberg method). isomiRs with a log2 fold change > 2 or < -2 and a p-adjusted < 0.05 were considered significant.
E2F activity score
E2F activity scores were calculated from the TCGA-BRCA and METABRIC mRNA data as follows: Genes present in the MSigDB Hallmark E2F target gene signature (version v7.3) were used. Expression of each gene was z-scaled over all patients. The median values of these z-scaled expression values were calculated for each patient over all genes of the E2F target gene signature; this median value of z-scaled expression of E2F target genes was used as the “E2F activity score” for each patient. Associations between E2F activity scores and expression of 5’isomiR-183-5p for each patient were depicted as scatter plots, and correlation coefficients were calculated using Spearman’s correlation test.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was applied to the mass spectrometry data. Briefly, missing values of mass spectrometry data were imputed and log2 fold changes of protein expression were computed for every protein and for all three 5’isomiR-183-5p compared to the control group. Protein identifiers were linked to the encoding genes for further downstream analyses. Gene set enrichment analysis was performed on the pre-ranked gene list using the Hallmark Gene Set Collection (version 7.3), a weighted enrichment statistic and default parameters of GSEA software [36–38]. The analyses were summarized in a bubble heatmap depicting the normalized enrichment score (NES) and the false discovery rate (q-value). To show individual GSEA graphs, we used an adapted version of the replotGSEA function from the Rtoolbox (https://github.com/PeeperLab/Rtoolbox) to re-arrange the output of the GSEA tool.
Gene set enrichment analysis of TNBC patients from TCGA and METABRIC datasets was performed to investigate the correlation between isomiR expression and the activity of the E2F and other pathways in these patients, again using the Hallmark Gene Set Collection [38]. To address this, batch-corrected expression of the respective isomiR (TCGA data) or microarray-based expression values for miR-183-5p (METABRIC data) were converted to ranks across patients and used as parameter. Ranked mRNA expression data were used as an input file. The collection of Hallmark gene sets was used [38]. Thereby, Spearman correlation coefficients between isomiR/miR and genes were used as a ranking metric and 1000 permutations were performed by phenotype to assess statistical significance. The results were visualized in a bubble heatmap depicting the normalized enrichment score (NES) and the false discovery rate (q-value).
Graphical illustration and statistical analysis
For analyses performed in R, we used R version 4.0.2 or 4.0.3 (R Core Team 2020. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/) and RStudio (RStudio Team, 2020. RStudio: Integrated Development Environment for R. RStudio, PBC, Boston, MA. URL http://www.rstudio.com/). Graphs were either generated using base R or using ggplot2 (version 3.3.3) [39] if not stated differently.
For heatmap visualization of the z-scaled mass spectrometry results, the pheatmap package (version 1.0.12) [40] was used. Distance measures between rows were calculated by the Pearson correlation coefficient and clusters were compared by complete linkage.
If not mentioned differently, data are presented as mean ± SD. Statistical analysis was performed by two-tailed Student’s t-test using GraphPad Prism (version 9.3.0) and p-values < 0.05 were considered statistically significant. p-value < 0.05, < 0.01 and < 0.001 are indicated with one, two and three asterisks respectively.
{kind=link}