Overview of viral genome in Lake Fuxian
From the five shotgun metagenomes and coassembly results, 1,464 viral sequences (> 10 kb) were recovered from Lake Fuxian. Of these sequences, 942 were identified by the de novo method (VirSorter), 211 showed high similarity with reported bacteriophage sequences in the IMG/VR database, and 311 were recovered by both methods (Additional File 1: Table S1). Both approaches for bacteriophage identification employed in this study (VirSorter and comparison to IMG/VR) were based on an a priori algorithm with known bacteriophage sequences and core bacteriophage proteins, reducing the number of FXLVGs detected but increasing the accuracy of the results [59, 60]. After clustering at least 95% sequence similarity and 85% aligned coverage, 985 nonredundant Lake Fuxian viral genomes (FXLVGs) were obtained for further analysis, with genome lengths ranging from 10 kb to 451.7 kb and guanine-cytosine (GC) contents ranging from 19.37% to 74.05% (Additional File 1: Table S1). Four bacteriophages measured more than 200 kb in length and were classified as prophages by VirSorter; therefore, we did not consider them as giant viruses that were reported in previous study [61]. FXLVG richness at 20 m in depth was the highest (three times larger than the average richness at other depths), while the richness from 60 m to the bottom decreased rapidly (Fig. 1a; Additional File 2: Table S2). The FXLVG richness status also supported the profile that not only microorganisms, including bacteria and archaea, but also phages had stratified features during the holomictic period [58].
The genomic heterogeneity of the freshwater virome dataset was evaluated in our study. FXLVGs were compared with viral data obtained from other freshwater habitats at the nucleic acid level with the same sequence similarity cutoff used in FXLVG construction. There was no FXLVG matching with either the UFO (Uncultured Freshwater Organisms) or LBVC (Lake Biwa Viral Contigs) viral datasets, indicating that the freshwater virome had high habitat heterogeneity. A total of 655 FXLVGs were novel sequences that were first reported in this study (Additional File 3: Fig. S1) [27, 55]. It might not be productive to compare viral genomes from similar freshwater habitats even with the same bacteriophage identification pipeline, let alone virome dataset from different habitats, such as TARA Ocean, the large ocean virome GOV2.0 dataset generated by at least three kinds of viral sequences detection software [7]. Remarkably, among 322 IMG/VR-like FXLVGs, 245 (76.09%) were from freshwater, and 59 (18.32%) were from other aquatic ecosystems, such as marine and wastewater (Additional File 4: Table S3). A total of 94.41% of IMG/VR-like FXLVGs were traced from aquatic environments, suggesting that our viral detection pipeline was reliable.
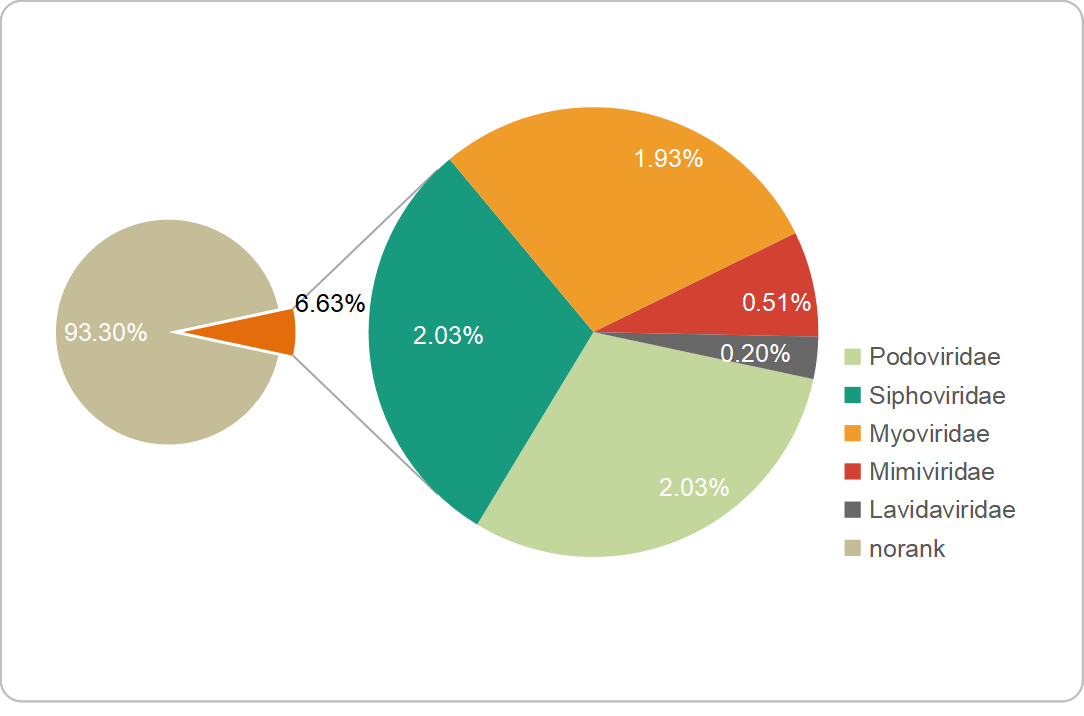
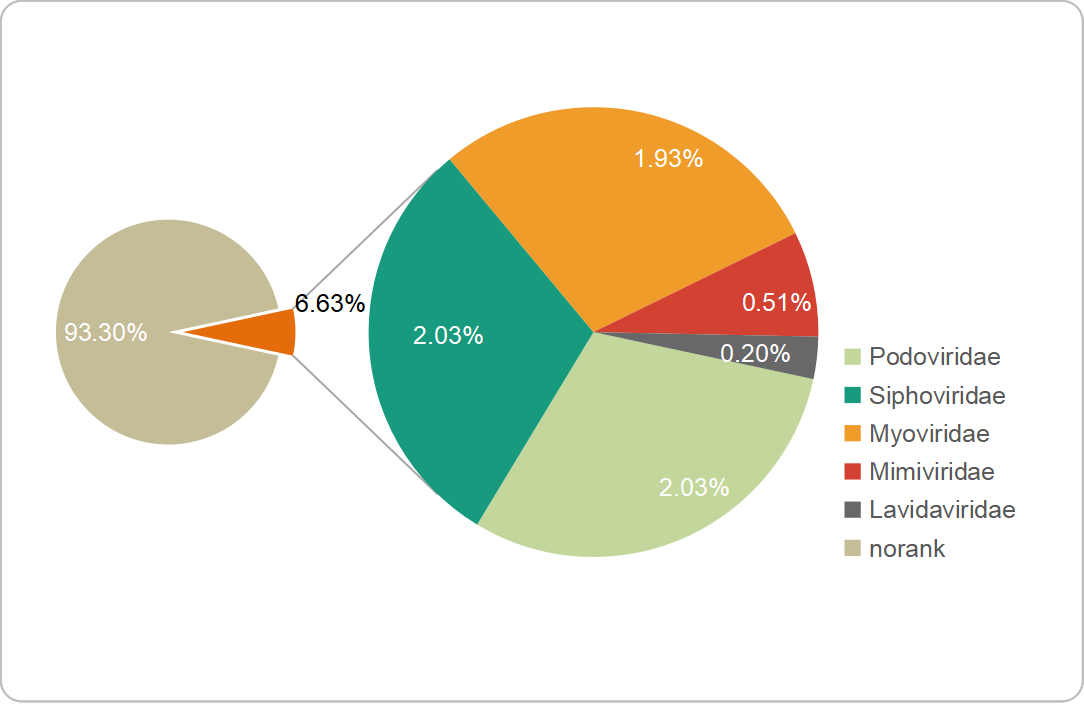
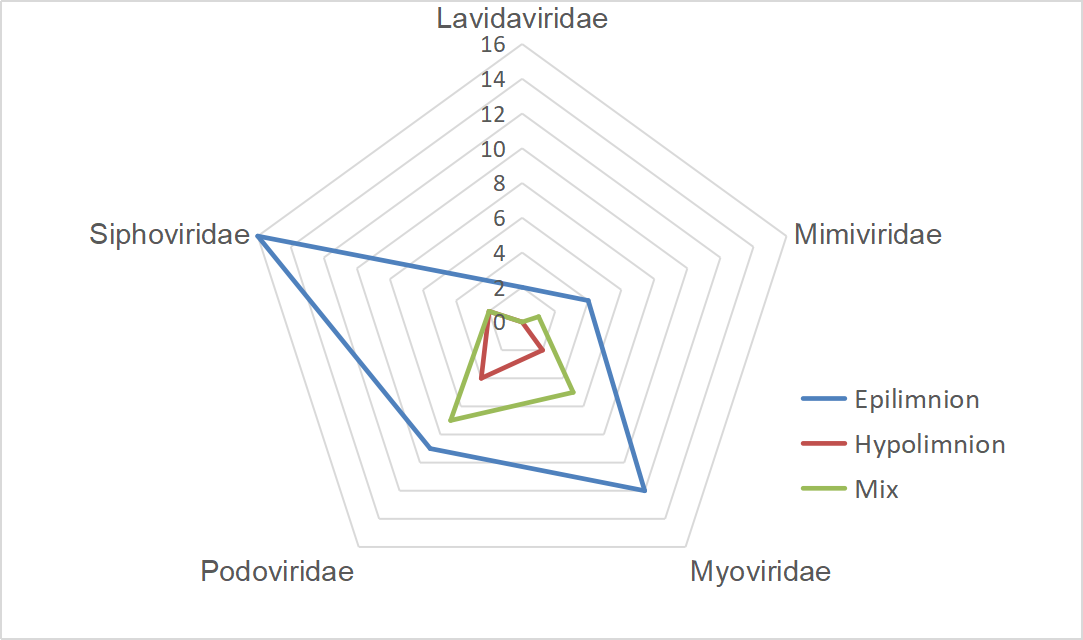
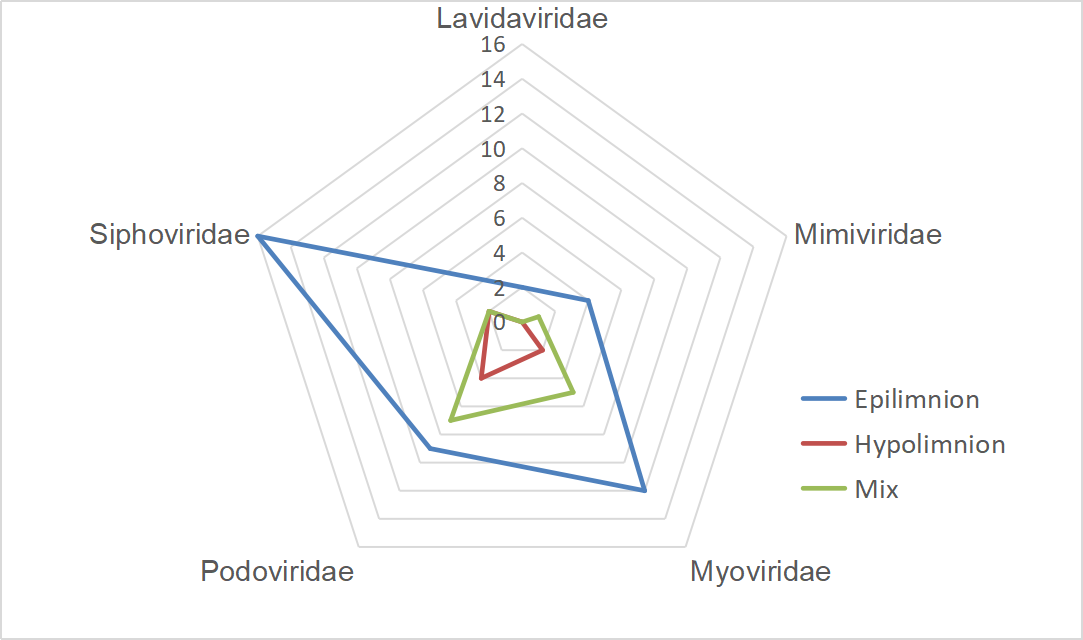
A gene-sharing network was constructed by vConTACT2 to classify FXLVGs (985 accessions) with publicly available taxonomy-known viral sequences in RefSeq (12,155 accessions, March 2020). The classification using vConTACT2 was able to cluster viral sequences with known genomes. A total of 57.44% (n=7547, 7547/13,140) of viral accessions were clustered, and the remaining accessions did not cluster with any viral genomes. Of the 985 FXLVGs, 778 could be assigned to 158 viral clusters (VCs), of which 21 VCs contained taxonomy-known viral genome members (Additional File 4: Table S3; Additional File 5: Fig. S2). Finally, 66 (6.63%) FXLVGs clustered within the VCs belonging to the families Podoviridae (n=20), Siphoviridae (n=20), Myoviridae (n=19), Mimiviridae (n=5) and Lavidaviridae (n=2). Except for five Mimiviridae FXLVGs, other known-family FXLVGs belong to Caudovirales classified as double-stranded DNA viruses (dsDNA viruses). Mimiviridae and Lavidaviridae were detected only in the epiliminion, while the ratio of Podoviridae in the hypolimnion was two times that in the epiliminion. These results indicated that the majority of the FXLVGs obtained from Lake Fuxian were completely unclassified, and the bacteriophage taxonomic structure showed stratification between the epiliminion and hypolimnion during the holomictic period (Additional File 6: Fig. S3). Interestingly, the bacteriophages whose taxonomic percentages were in the top three were Podoviridae, Siphoviridae and Myoviridae in Lake Fuxian, which coincided with those identified from the ocean virome GOV2.0 and wastewater viromes [7, 18]. In Lake Fuxian, putative hosts for Myoviridae, Podoviridae and Siphoviridae were members of Caulobacter, Myxococcus and Enterobacteria. In wastewater plants, the most abundant hosts were Acinetobacter, Arcobacter, and Moraxella [18]. This finding demonstrated that host structure could be more diverse than viral taxonomy structure among different habitats.
Depth profile for bacteriophage abundance and gene function
The relative abundance of FXLVGs was determined based on the metagenomic average depth, calculated as read coverage per base under the same read input for each sample. Among the 985 FXLVGs, 695 (70.56%) were epilimnion-specific, and 161 (16.35%) were hypolimnion-specific, which was consistent with the viral richness results (Fig. 1b). With respect to the GC content of the clean metagenomic reads, an increasing GC content of FXLVGs was observed between the epi- and hypolimnion (40.75%±9.09% and 51.98%±12.31%), whose pattern coincided with those of MAGs (Fig. 1c; Additional File 1: Table S1) [58]. In the hypolimnion, 17 FXLVGs were depth-specific (7 at 120 m, 6 at 140 m, 2 at 60 m and 2 at 80 m). These numbers were considerably lower than those observed at 20 m (epilimnion-specific FXLVGs). We observed that epilimnion-specific MAGs (higher-temperature habitats) had considerably smaller genome sizes than hypolimnion-specific MAGs (Additional File 7: Fig. S4). However, no depth profile was detected for FXLVG genome length based on the current genome completeness. Bacteria and archaea growing in a high-optimum-temperature environment were found to have smaller genomes in a previous report [62]. These results demonstrated that the genome evolutionary process might differ between bacteriophages and their prokaryotic hosts.
To further explore how bacteriophages might affect the biogeochemistry and the potential relationship between viral abundance specificity and function at different lake depths, AMGs in FXLVGs, which are host metabolic genes carried by viral genomes and function during the infection process, were categorized [63]. Overall, 567 AMGs were detected in 239 (24.26%) FXLVGs (2.37 AMGs per bacteriophage) (Fig. 2; Additional File 8: Table S4). Similar to previous freshwater and soil virome studies, carbohydrate metabolism-related AMGs were the most popular in FXLVGs [9, 12]. The proportion of AMG-encoding epilimnion-specific FXLVGs (198/695, 28.49%) was significantly higher than that of hypolimnion-specific FXLVGs (18/161, 11.18%) (Fisher’s exact test, p < 0.01). The proportion differences of aromatic compounds, glycans, nucleotides and sulfur relay metabolism-related AMGs between epi- and hypolimnion-specific FXLVGs exceeded more than 2-fold (Fig. 2). Among the four metabolism categories, epilimnion-specific FXLVGs exhibited more AMGs in aromatic compound and glycan metabolism, while hypolimnion-specific FXLVGs exhibited more AMGs in nucleotide and sulfur relay metabolism.
Notably, there were two AMGs involved in sulfur relay metabolism, moeB (molybdopterin-synthase adenylyltransferase, EC:2.7.7.80) and mec ([CysO sulfur-carrier protein]-S-L-cysteine hydrolase, EC:3.13.1.6). Both genes were detected in epi- and hypolimnion-specific FXLVGs, indicating that bacteriophages might universally be involved in the sulfur relay cycle in the host genome. Moreover, the proportional differences in amino acid, carbohydrate, cofactor/vitamin, energy, lipid, terpenoid/polyketide metabolism and secondary metabolite-related AMGs between epi- and hypolimnion-specific FXLVGs were minor (< 2-fold difference) (Fig. 2). However, regarding the detailed gene functions, there were large differences. For example, the glycine hydroxymethyltransferase (glyA, EC:2.1.2.1) gene involved in methane metabolism only existed in epilimnion-specific FXLVGs. In contrast, pdhB (pyruvate dehydrogenase E1 component beta subunit) and aceF (pyruvate dehydrogenase E2 component), two members of the citrate cycle (TCA cycle), were only detected in the hypolimnion-specific FXLVG867 (Additional File 8: Table S4). These results demonstrated that bacteriophages might play important roles in basic carbohydrate metabolism in deep freshwater ecosystems.
All these results demonstrated that the epi- and hypolimnion-specific lineages were clearly differentiated by viral abundance and GC content, and depth-specific AMGs might engage in favorable functional niches.
CRISPR-based bacteriophage-host link events
Since the introduction of new bioinformatic tools in the data analysis pipeline, the MAG dataset in this study has improved considerably compared with a previous report [58]. Coassembly and binning of metagenomes initially generated 2,431 metagenome-assembled genomes (FXLMGs) for Lake Fuxian, including 405 high-quality (> 90% completeness and < 5% contamination), 578 medium-quality (≥ 50% completeness and < 10% contamination) and 1,448 low-quality (others) FXLMGs, doubling the number of FXLMGs in the former dataset (Additional File 9: Table S5). After clustering at 95% ANI, 431 high- or medium-quality representative FXLMGs, including diverse bacterial and archaeal phyla, remained (Additional File 10: Table S6). Among 431 representative FXLMGs, 423 (98.14%) were bacterial FXLMGs spanning 22 phyla, where Proteobacteria (n = 134), Actinobacteriota (n = 71), Planctomycetota (n = 51), Verrucomicrobiota (n = 34), Bacteroidota (n = 30) and Patescibacteria (n = 22) were highly represented (> 5%). Within the domain Archaea, 8 FXLMGs, Crenarchaeota (n = 2) and Nanoarchaeota (n = 6), were recovered (Fig. 3).
As a relatively reliable computational method for detecting bacteriophage-host association signals, the CRISPR spacer-based approach has been applied in many virome studies [10, 14, 26-28]. A total of 7,075 CRISPR spacer sequences were identified from 393 representative MAGs covering a total of 24 phyla (Additional File 10: Table S6). The spacer sequencer distribution at the phylum level was similar to the FXLMG numbers across each phylum. The top five phyla (Proteobacteria, Actinobacteriota, Planctomycetota, Verrucomicrobiota and Bacteroidota) also had the highest numbers of spacer sequencers (Fig. 3). The spacer sequence number varied notably at the single-FXLMG level; for example, one Myxococcota FXLMG had 526 spacer sequences, while 49 FXLMGs only had 1 for each (Fig. 3; Additional File 10: Table S6; Additional File 11: Fig. S5). Furthermore, only 10 CRISPR-based virus-host events (10 FXLVGs and 8 FXLMGs) were identified from 985 FXLVGs detected in Lake Fuxian (Additional File 12: Table S7). Among these sequences, one Chloroflexota FXLMG (FXLMG137) linked with three FXLMGs, but the other FXLMGs linked one-by-one with a unique FXLVG. We also searched CRISPR spacer sequences in unbinned or no-FXLVG fractions. A total of 2,895 spacer sequences and 19 link events were recovered, but only 10 hosts could be classified as bacteria, while the others were unclassified (Additional File 13: Table S8).
Similarly, in cone pool virome research, only three bacteriophage-host link events were detected by CRISPR [10]. Twenty-six (1.28%, 26/2034 total viral genomes) bacteriophages had host information identified by the CRISPR-based method in two freshwater habitats [27]. These results suggested that most MAGs lack a CRISPR defense system to combat current phages, and existing CRISPR elements across the host genomes might be evolutionary traces of host bacteriophage-fighting machinery in the history of host genomes [28]. On the other hand, the bioinformatic principle of FXLMG reconstruction could not be constructed efficiently for very low-abundance microbial genomes [6, 22]. Meanwhile, bacteriophages could also control host abundance to an extent. Nevertheless, the low efficiency of CRISPR-based link event detection in Lake Fuxian suggests the need to use other strategies for linking bacteriophages to their hosts.
Hi-C-based bacteriophage-host link events
Because of the highest richness and abundance of FXLVGs in the epilimnion, we chose the epilimnion sample as a model to investigate the bacteriophage-host relationship by Hi-C technology. A total of 3,118 candidate bacteriophage-host link events were initially identified (Additional File 14: Table S9). Considering the bacteriophage and host abundance and the number of restriction sites in each sequence, 58 link events (confidence > 95%) were eventually detected across 31 representative FXLMGs spanning 8 phyla (Fig. 3). Among 58 link events, 8 FXLVGs had more than one host, and 2 of them might infect FXLMGs across different phyla. In addition, 11 FXLMGs could be infected by multiple FXLVGs (Additional File 15: Fig. S6). There were 24 (24/58, 41.38%) events (among 8 bacteriophage-host clusters), where the corresponding FXLVGs contained AMGs (Fig. 4a). Interestingly, link events detected by CRISPR and Hi-C were completely different in our study. The low number of CRISPR-based events decreased the possibility of overlap event detection, and the Hi-C-based method was focused on the infection events within each host cell in situ. Therefore, it was reasonable that there was no link event detected by either strategy.
Strong correlation signals for functional complementarity between FXLVGs and FXLMGs were observed in the Hi-C-based results. The infection signals of 11 Cyanobacteriota-FXLVG link events were identified, including 2 epilimnion-specific FXLMGs and 8 epilimnion-specific FXLVGs (Additional File 12: Table S7). Four of the eight FXLVGs (FXLVG66, FXLVG309, FXLVG472 and FXLVG500) contained D1 or D2 proteins, which were members of the photosystem II P680 reaction center (psbA and psbD in the VIBRANT software classification system). Among these four FXLVGs, proteins PC and Fd, which participate in photosynthetic electron transport, were also identified (Additional File 16: Fig. S7). Remarkably, one of 8 cyanophages, FXLVG608, had the mec gene ([CysO sulfur-carrier protein]-S-L-cysteine hydrolase, EC:3.13.1.6), and its infected host was FXLMG141, which lacked the mec gene in the sulfur relay system (Fig. 4b). These results indicated that cyanophages might provide other important genes to Cyanobacteriota in addition to the most widely known D1 and D2. In previous reports, D1/D2 proteins in viral AMGs were generally used to predict potential Cyanobacteriota hosts [26, 27, 64, 65], but the limitation was that only phylum information of the host could be predicted, and the corresponding bacteriophages were uniformly classified as cyanophages.
Another bacteriophage-host functional complementary event was found in the mannose-fucose metabolism pathway (KEGG ko00051) but with different strategies of cooperation. Mannose and fucose are sugars, with the former being an essential endoplasmic reticulum component in prokaryotes, while the latter is distributed in complex carbohydrates of all species. In prokaryotes, GDP fucose is synthesized from GDP-mannose in a three-step reaction catalyzed by two enzymes, GMDS (GDP-mannose 4,6-dehydratase, EC:4.2.1.47) and TSTA3 (GDP-L-fucose synthase, EC:1.1.1.271) (Fig. 4c) [66]. In the FXLVG611-FXLMG349-FXLMG350 cluster (VHC4), two key enzymes (GMDS and TSTA3) were identified in the bacteriophage, and both hosts contained a PPM gene (phosphomannomutase, EC:5.4.2.8), which was used to transform mannose-6-phosphate into mannose-1-phosphate. In this case, it appears that the host could utilize the fucose synthesized within the bacteriophage. However, in the FXLVG794-FXLMG254 cluster (VHC5), only TSTA3 was identified in FXLVG794, and GMDS was observed in the host FXLMG254 (Planctomycetota). These results demonstrated that bacteriophages might play an important role in fucose synthesis, which is necessary in bacterial cell construction, although cooperation can be very diverse. Nevertheless, these findings demonstrate the considerable power of Hi-C in bacteriophage-host link event detection in the future.
Bacteriophage-host interactive abundance analysis
Based on the identified bacteriophage-host pairs and genome coverage information obtained by the read mapping approach along the depth profile, this research provided an opportunity to evaluate the abundance relationship between bacteriophages and hosts at species level. For 58 Hi-C-based bacteriophage-host pairs in five samples (290 events), 170 (58.72%) events were discarded in further analysis because of undetectable coverage of the bacteriophage and/or host genome (Additional File 17: Table S10). All 170 events were from hypolimnion samples, which might be observed because only the epilimnion sample was selected to perform the Hi-C experiment, and no hypolimnion-specific FXLVG was identified in the Hi-C-based bacteriophage-host relationship results. Among the remaining 120 events, more than half of the hosts (62/120, 51.67%) had higher genome coverage than their bacteriophages (bacteriophage/host abundance ratios, BHR < 1.5). A total of 22.5% (27/120) of events had nearly equal genome coverages (-1.5 < BHR < 1.5), and 25.83% (31/120) had higher bacteriophage coverage (Additional File 18: Fig. S8). Notably, these results were significantly different from those obtained in a previous virome study in which most bacteriophages had greater abundance than their hosts [10, 67]. The uncertainty provides another opportunity to use the Hi-C-based method to detect the bacteriophage-host relationship within prokaryotic cells instead of counting bacteriophages at lysis time.
Similar to FXLVG abundance, BHR showed a specific pattern along the depth profile and host diversity. BHRs were significantly (p < 0.01) higher in the epilimnion than in the hypolimnion (Fig. 5a). The BHRs in the epilimnion and hypolimnion were both negatively correlated with host abundance (Fig. 5b), and BHRs in the epilimnion decreased considerably more quickly with lower host abundance. Remarkably, BHRs varied among different hosts at the phylum level (Additional File 19: Fig. S9). For Cyanobacteriota, total FXLVGs had a higher abundance than FXLMGs (> 6.97-fold on average). For Planctomycetota, FXLMGs in hypolimnion had higher abundance where the corresponding BHRs were in considerably lower abundance, while in epilimnion, the opposite distribution was observed. These results were in keeping with the “Piggyback-the-Winner” theory that lysogeny was the predominant viral lifestyle in a high-host-abundance environment where bacteriophages (integrated into the host genome or coexisting within the host cell) increased the ability of the host to resist other bacteriophage infections and gain competitive advantages compared with other microbes [68].
{kind=link}
{kind=link}
{kind=link}
{kind=link}