Bacterial strains, plasmids, and growth conditions
B. glumae strains and plasmids used in this study are listed in Table 2. All strains were stored at −80 °C in 20% glycerol and grown in nutrient broth (NB) medium (1% peptone, 0.3% yeast extract, and 0.5% NaCl, pH 7.5) at 30 °C. Luria-Bertani (LB) agar without sucrose and LB agar containing 10% (w/v) sucrose were used for deletion mutagenesis. Antibiotics were added at the following concentrations when required: 50 µg/ml kanamycin (Km) and 100 µg/ml ampicillin (Amp).
Table 2
Strains and plasmids used in this study.
| Strain or plasmid | Relevant characteristicsa | Source or reference |
| Burkholderia glumae | | |
| AU6208 | Human pathogen isolated from surgical specimens from lung lesions in a 8-month-old boy | Weinberg et al., 2007 |
| LMG 2196T | Isolated from Oryza sativa, grain | Shannon Johnson, 1967 |
| AU6208ΔBCAL3235 | AU6208 deletion mutation defective in BCAL3235 | This study |
| AU6208ΔSbnAB | AU6208 deletion mutation defective in SbnAB | This study |
| AU6208ΔpmlR | AU6208 deletion mutation defective in pmlR | This study |
| Escherichia coli | | |
| DH5α | F− Φ80d lacZΔM15Δ(lacZYA-argF) U169 recA1 endA1, hsdR17(rk−, mk+) phoA supE44λ− thi-1 gyrA96 relA1 | This study |
| S17-1(λ pir) | λ Lysogenic S17-1 derivative producing π protein for replication of plasmids carrying oriR6K; recAprohsdRRP4-2-Tc::Mu-Km::Tn7 λ− pir | Simon et al., 1983 |
| BL21(DE3) | F− ompT hsdS20(rb−, mb−) gal | Novagen |
| Plasmids | | |
| pGEM-T | AmpR; cloning vector | Promega |
| pKMS1 | KmR; R6K-based suicide vector; requires the pir-encoded π protein for replication | Zou et al., 2011 |
| pKMS1- BCAL3235 | KmR; Used to create knockout mutant of AU6208ΔBCAL3235 | This study |
| pKMS1-SbnAB | KmR; Used to create knockout mutant of AU6208ΔSbnAB | This study |
| pKMS1-pmlR | KmR; Used to create knockout mutant of AU6208ΔpmlR | This study |
| aKmR and AmpR indicate kanamycin and ampicillin resistant |
DNA extraction, genome sequencing, and genomic analysis
Bacterial genomic DNA was extracted using TIANamp Bacteria DNA kits (Tiangen Biotech, Beijing, China). Whole-genome DNA sequencing of B. glumae AU6208 was conducted using the Pacific Biosciences (PacBio) RSII Single Molecule Real Time (SMRT) system and Illumina with the NEBNext Ultra DNA Library Prep Kit (New England Biolabs, MA, USA). The combination of PacBio and Illumina sequence shotgun data was assembled with the SMRT analysis packages (v2.1) using hierarchical genome assembly (Chan et al., 2014; Lau et al., 2014). Low-quality sequences were filtered by Sickle (v1.33) with Q30 (Joshi and Fass, 2011). Protein-coding regions, tRNAs, and rRNAs were predicted using rapid annotation subsystem technology[24]. Hits were then analyzed for signal peptide characteristics using SignalP [25]. Transmembrane domains and functional protein domains were analyzed using tied-mixture hidden Markov modeling and InterProScan, respectively [26, 27]. The secondary metabolite clusters, antibiotic resistance clusters and toxins, and virulence factors were predicted by comparing with DoBISCUIT, CARD, and VFDB databases, respectively [28–30]. The category of functional proteins was further annotated by Clusters of Orthologous Groups (COG) and Gene Ontology databases [31]. Genome comparison between AU6208 and LMG 2196T was conducted using average nucleotide identity (ANI) and alignment fraction (AF), which were calculated by ANIcalculator [32]. Synteny of genome structure between AU6208 and LMG 2196T was performed by Mummer 3.0 with default parameters [33].
Construction of deletion mutants and complementation
To investigate the role of genes of interest in AU6208, we constructed deletion mutants in AU6208 sing homologous recombination. Briefly, two fragments flanking the left and right borders of corresponding genes were amplified from the wild-type genomic DNA with the primer pairs listed in Table S5. The amplified fragments were cloned into pGEM-T (Promega), confirmed by sequence analysis, and then digested and subcloned into vector pKMS1 [34] at BamHI and PstI (or SalI) sites. The resulting recombinant plasmids were introduced into AU6208 by electroporation, and transformants were plated on NAN plates supplemented with kanamycin. Colonies resulting from a single homologous crossover (integration of the deletion construct at either the left or right border of the target gene) were then transferred to NBN broth, grown for 12 h at 28 °C, and then plated on NAS plates for sucrose-positive deletion mutant selection. Sucrose-resistant colonies were visible within 3 to 4 days and then transferred to NA plates and NA plus kanamycin plates. Since kanamycin-sensitive colonies could be mutants containing a second homologous crossover, these were further examined by PCR amplification.
In order to complement the deletion mutants, the full-length of corresponding genes were amplified using primer pairs listed in Table S6. After confirmation by sequence analysis, the amplified DNA fragments were cloned into pUFR034 at the BamHI and SalI sites to create the recombinant plasmids, the recombinant plasmids were transferred into corresponding mutants by electroporation, and transformants were screened on NA plates with kanamycin. A putative transformant was shown to contain recombinant plasmids by PCR analysis.
Horizontal gene transfer (HGT) analysis
Screening for HGT was first performed by HGTfinder with default parameters [35]. Confirmation of HGT was performed by a combination of BLAST screening and large-scale phylogenetic trees. BLAST screening was performed based on our previous study [36]. In brief, candidate sequences in B. glumae were compared against sequences in the NCBI reference genome using BLASTp [37] followed by extracting the sequences with highest similarity for further study. We considered two genes orthologs when the E-value < 10−10 and when the alignment similarity was higher than 30% with more than 85% coverage [38]. These sequences were aligned using MAFFT [39], and the conserved region of each alignment was trimmed using TrimAI [40] with stringent settings. Maximum likelihood and Bayesian phylogenies were generated based on our previous study [36].
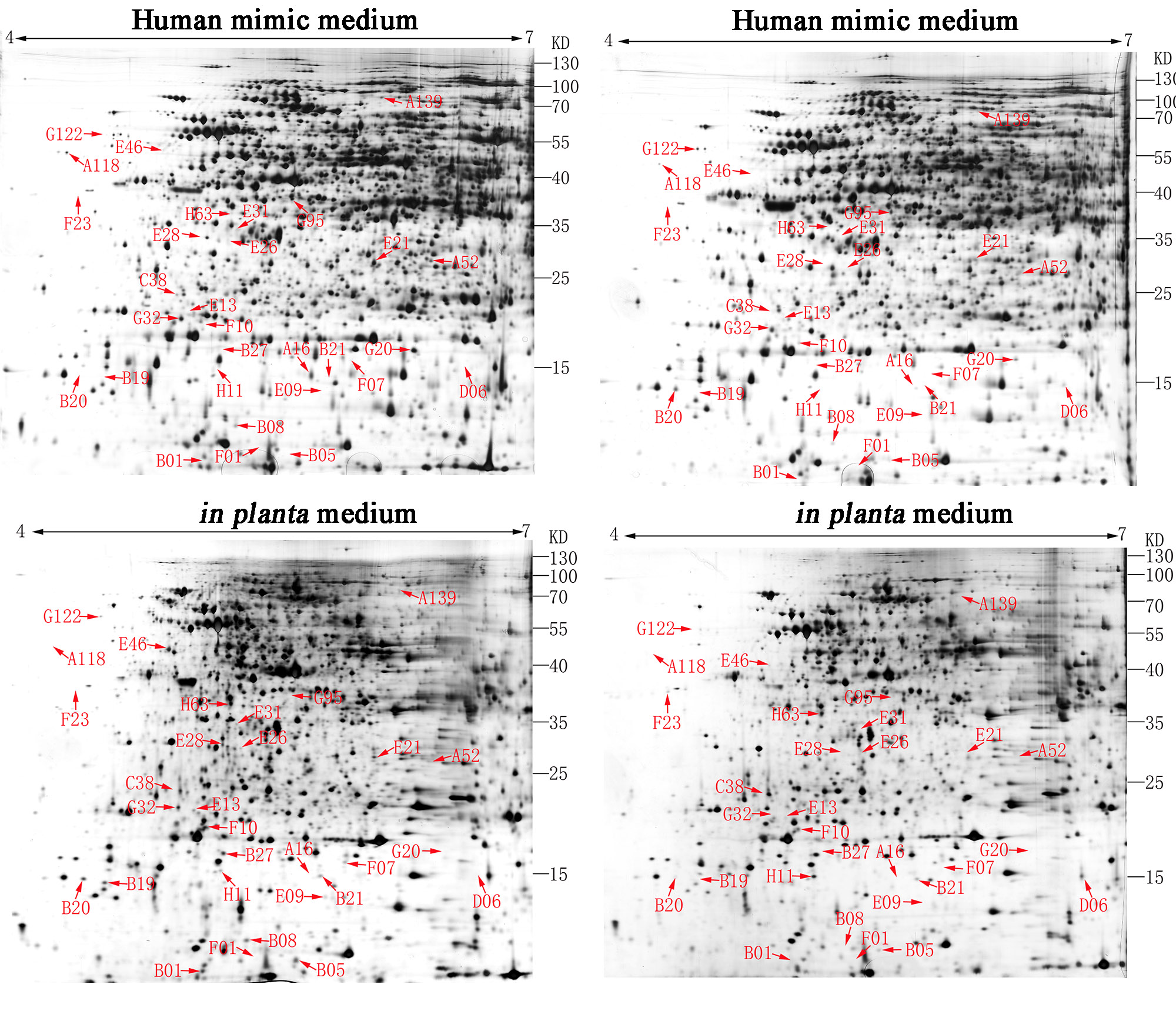
Preparation of bacterial samples from different niches for 2-dimensional electrophoresis
For the human mimic condition, overnight bacterial cultures from one single colony in NB broth were trans-cultured as 1:100 in 100 ml fresh SCFM2 (Human mimic medium) [22] and incubated with shaking at 200 rpm at 30 °C until the OD600 reached 0.8. Cells were then collected by centrifugation at 5,000 × g for 10 min, washed with phosphate-buffered saline (PBS) three times, and used for protein extraction. For the in planta mimic condition, we generally used the previous method [23]. In brief. the rice leaves were collected and homogenized by liquid nitrogen with a mortar and pestle. The resultant was aliquoted to 1 g in tubes and stored at − 80 °C before using. 40 mL culture of B. glumae was grown to the mid-exponential phase (A600 of 0.5) and 1 g resultant that was previous kept at − 80 °C was added to the culture medium. The bacterial pellets from 1.5 mL samples were immediately harvested by centrifugation at 5,000 × g for 20 min, washed with PBS three times, and used for protein extraction. Extraction of total proteins and 2-dimensional electrophoresis were performed as previously described [41]. The resulting protein profiles were analyzed with ImageMaster 2D platinum, version 5.0 (Amersham Biosciences, Uppsala, Sweden).
Protein in-gel digestion and liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
The tryptic in-gel digestion protocol, optimized for a polyacrylamide gel of 1.5-mm thickness, was performed based on a previous method [42]. Briefly, protein spots were excised from the gel and put into the 96-well polypropylene ZipPlate Micro-SPE plate (Thermo). Then, the gel piece was rinsed for 40 min with 100 µl of 25 mM ammonium bicarbonate/acetonitrile (95:5, v/v) followed by two rinses with 100 µl of 25 mM ammonium bicarbonate/acetonitrile (50:50, v/v). The gel piece was then dehydrated for 12 min with 200 µl of acetonitrile. Digestion was performed with 15 µL of sequencing grade porcine trypsin (10 ng µl− 1) at 37 °C for 3 h.
The peptides released from trypsin digestion for LC-MS/MS analysis were prepared in two biological replicates. LC was performed using a Dionex Ultimate 3000 nano-LC system following the method in our previous paper [43]. The MASCOT LC-MS/MS ion search algorithm (Matrix Sciences) was applied to evaluate the LC/MS spectra, and the sequence similarity of resulting peptides to the B. glumae AU6208 strain was compared to other Burkholderia species accessible on NCBI. To further identify the proteins, the cross-correlation (X corr) scores of singly, doubly, and triply charged peptides were fixed greater than 1.8, 2.5, and 3.5, respectively [43]. Peptide sequences with the highest X corr values were then identified.
Quantitative real-time PCR
Total RNAs were extracted from exponentially growing cells, using an RNeasy Mini spin columns Kit (Qiagen) and was treated with a unit of RNase-free DNase I (Qiagen), and cDNA synthesis was performed with a Moloney murine leukemia virus reverse transcriptase first-strand cDNA synthesis kit (QIAGEN). The cDNA was then used directly as the template for qRT-PCR using a SYBER Green master mix (Protech Technology Enterprise Co., Ltd.) on an ABI Prism 7000 sequence detection system (Applied Biosystems). Primers for quantitative real-time PCR (qRT-PCR) of the selected genes were designed by using Primer 3 [44] and gyrB was used as internal control. Ct values were shown on Table S6.
Pathogenicity assay
In a greenhouse, rice plants (Oryza sativa cv. kitaake) were inoculated at the flowering stage with approximately 1 × 109 CFU/ml of B. glumae. Disease symptoms in the rice were evaluated on day 10 after inoculation. The disease index was determined as described previously [45]. In detail, 0 = healthy panicle, 1 = panicle 0 to 20% discolored, 2 = panicle 20 to 40% discolored, 3 = panicle 40 to 60% discolored, 4 = panicle 60 to 80% discolored, and 5_= panicle 80 to 100% discolored [disease index = Σ(number of samples per score X score)/the total number of panicles]. Pathogenicity assays were repeated three times with four replicates.
Biofilm formation and exopolysaccharide (EPS) secretion assay
Biofilm formation was determined using a previously described method [46]. In brief, bacterial cells grown overnight were inoculated 1:100 in fresh NB media in a 96-well microplate (Corning-Costar Corp., Corning, NY, USA). After static culture at 30 °C for 48 h, cells were stained with 1% crystal violet (CV) for 15 min. The planktonic cells were removed by several rinses with H2O. The CV-stained bound cells were air-dried for 1 h and dissolved in 90% ethanol, and the optical density at 590 nm (OD590) of the solution was measured to quantify biofilm formation.
The extraction and quantitation of bacterial EPS was performed as described previously [47]. Briefly, overnight bacterial cultures were inoculated 1:100 in 100 ml fresh NB media to OD600 = 1.0. Cultures were centrifuged at 4 °C, and 10 µl of the supernatant was removed and filtered through 0.22-µm Millipore filters to remove all bacterial cells. A mixture of one volume of supernatant with four volumes of Sevage’s solution (4:1 chloroform:butanol) was incubated with shaking at 180 rpm for 30 min. Subsequently, the supernatants were collected by centrifuging and then mixed with three volumes of 95% ethanol and incubated at 4 °C for 24 h without shaking. The pellet was harvested, dried, and weighed after incubation. The experiments consisted of three biological replicates and were repeated at least three times.
Statistical analysis
STATGRAPHICS Plus, version 4.0 (Copyright Manugistics Inc., Rockville, MD, USA) was used to perform the statistical analysis. Levels of significance (p < 0.05) of main treatments and their interactions were calculated by analysis of variance after testing for normality and variance homogeneity.
{kind=link}