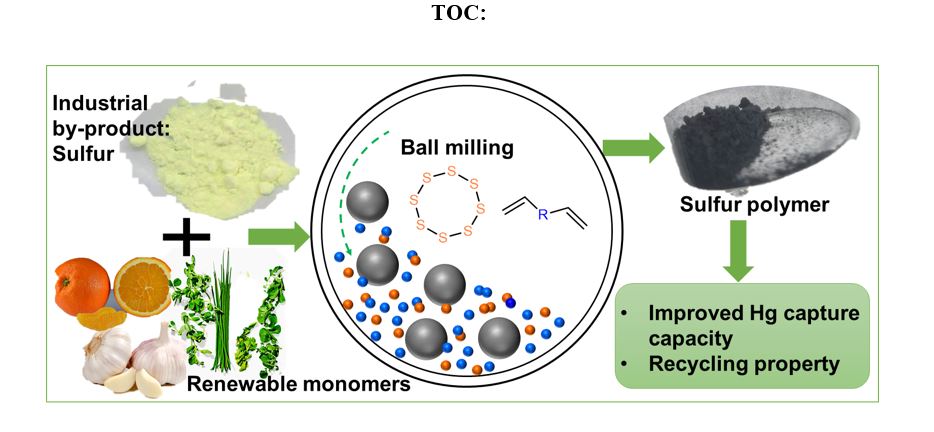
We synthesized 9 polymers using a ball mill, starting from different crosslinkers ranging from aromatic, aliphatic to volatile monomers including 1,3-diisopropenylbenzene (DIB), dicyclopentadiene (DCPD), divinylbenzene (DVB), 5-ethylidene-2-norbornene (ENB), limonene, myrcene, dially disulfide (DADS), styrene and isoprene (Fig. 1). The synthetic polymers were named as MS(S-monomer) respectively. In addition, 8 polymers derived from the same monomers, omitting isoprene which is unable to be used at high temperature, were synthesized using the conventional thermal synthesis (TS) method to act as control group, and were named as TS(S-monomer) respectively. The experimental procedures of the materials synthesis can be found in the supporting information (SI). MS(S-DIB) and MS(S-Styrene) were used as model reactions to optimize the reaction procedure, as DIB is a well-known monomer, which can chemically stabilize polymeric sulfur, and styrene is a monomer with lower stabilizing efficiency for sulfur because of the lower number of reactive bonds relative to the molecular mass, and linear resulting structure. As differential scanning calorimetry (DSC) curves in Fig. S1 show, the obtained polymer MS(S-DIB) shows a clear glass-rubber transition even after only 1-hour of reaction, and a clear increase of glass transition temperature (Tg) from -11°C to -1°C with the reaction time increase from 1 hour to 3 hours, suggesting that DIB can react with sulfur quickly to form sulfur-polymer by ball milling, and further crosslinking reaction occurs during extended reaction time. However, MS(S-Styrene) still has an obvious melt peak of unreacted crystalline sulfur even after 2-hours reaction, and there is only a slight glass-rubber transition apparent. After reacting for 3 hours, there is no unreacted crystalline sulfur remaining in polymerization system, and the product has a very clear glass-rubber transition. That indicates that styrene needs longer time to fully react with sulfur compared with DIB, and is able to form a sulfur polymer after 3-hours reaction.
Hence, based on the results of optimisation experiments, all other polymers were synthesized for 3 hours in order to fully convert the S8. To be clear here, all the characterizations of the products were carried out once the polymers were formed without any further treatment unless there is a special illustration. The DSC curves of all the polymers (Fig. S2) show that every polymer has a clear Tg and almost all polymers have no unreacted crystalline sulfur remaining except a slight sulfur melt peak in MS(S-Myrcene) after 3-hours polymerization. It’s worth noting that the volatile monomer isoprene (b.p. 34°C) was proven to successfully react with sulfur to form sulfur polymer by MS method here, which definitely cannot be realised through the conventional thermal synthesis process. Comparing the Tg of MS products with that of TS products (Fig. S3), it is interestingly found that some of the MS products have much lower Tg than the relevant TS products synthesized from the crosslinkers including DIB, DCPD, DVB, ENB and DADS, which would form crosslinked network normally, but other MS products formed from monomers including myrcene, limonene and styrene, show slightly higher Tg than the relevant TS products which are tend to be formed as linear polymer normally. From the powder x-ray diffraction (PXRD) curves of the MS products in Fig. S4, we can see that there is a trace of crystalline sulfur persisting in some of the polymers although it is not apparent from the second heating cycle in DSC curves. The unreacted sulfur can be removed by Soxhlet extraction in acetone as evidenced by the absence of peaks in PXRD patterns (Fig. S5a) of the MS products after Soxhlet extraction, but it needs to be mentioned that further reaction could occur during heating resulting from the dynamic disulfide bonds, since the DSC results (Fig. S5b) which show an increase of Tg of all the polymers after Soxhlet extraction. Hence, all the characterizations of MS polymers were done directly after 3-hours reaction in the ball mill to investigate their true chemical and physical properties and applications. According to solubility evaluation results in Fig. S6, all the MS polymers were demonstrated to be insoluble in neither tetrahydrofuran (THF), chloroform or acetone, which is notable. As Fig. S7 shows, TS(S-Myrcene), TS(S-Limonene), and TS(S-Styrene), which either exhibit linear or branched rather than fully crosslinked structures, are fully soluble in THF and chloroform and partly soluble in acetone. Other TS polymers, which are more highly crosslinked, are insoluble or only partly soluble in those three solvents. Theoretically, higher crosslinking degree will give higher Tg and higher solvent resistance to the polymer. Here one question arises, why should some of the MS polymers possess lower Tg than the TS polymers, but show higher solvent resistance? Elemental analysis results in Table S2 shows that most of obtained polymers contain high sulfur content but there are changes on the ratios of C:H and C:S after polymerization. The most suspected way which can cause elemental ratio change is the side reaction—H2S gas generation in thermal synthesis, which can cause C:H ratio and C:S ratio increase as elements S and H are lost. However, the C:H ratio and C:S ratio here both decreased after MS. So, another question comes up—how this happened? Both questions suggest there might be something different from MS method to the normal TS method in mechanism.
In order to investigate these questions and better understand the nature of the chemical reaction in the ball mill, styrene was chosen as model monomer to monitor the reaction as it theoretically has the least reactive activation point. Fourier transform infrared spectroscopy (FT-IR), hydrogen nuclear magnetic resonance spectroscopy (1H NMR), and X-ray photoelectron spectroscopy (XPS) were used to monitor the chemical environment changes as a function of reaction time. From FT-IR curves in Fig. 2A, it is observed that the peak at ~1626 cm−1 belonging to the C=C stretch and peaks at ~989 cm−1 and ~906 cm−1 belonging to C=C bend both disappeared with the increase of the reaction time, which suggests that vinyl groups in styrene were consumed to form polymer. Meanwhile, there is no change of the peaks belonging to the benzene ring, suggesting that there is no reaction occurring between the benzine group and sulfur, but there is a new peak at ~1701 cm−1 appearing after reaction, which is attributed to a C=O group. The same results can be observed from the FT-IR spectra of TS(S-Styrene) (Fig. S8). In addition, DSC curve (Fig. S9) of the control experiment MS(S-Toluene) suggests that there is no reaction between sulfur with saturated hydrocarbon. Due to insoluble property of the MS(S-Styrene), it is difficult to obtain the structure information of the produced polymer using solution NMR, but it is clear that the monomer styrene is fully consumed after 3-hours reaction from the 1H NMR spectra (Figs. S10 and S11) of monomer and products, resulting from the disappearance of the peaks belonging to hydrogen protons of styrene. In addition, energy dispersive spectroscopy (EDS) was used to detect the elements existing in the polymers. It is surprisingly found that there is Fe present in addition to the elements S and C in the polymer from the EDS images of MS(S-DIB) in Fig. 2B. That indicates that there are some iron extracted from steel balls trapped in the polymers, and there is a trace of Cr associated with Fe existing in the polymer, which is normally used in steel production. Moreover, Fig. S12 indicates that Fe was not uniformly dispersed in the polymer. In this case, anchoring of the polymeric products to Fe was considered to be the probable reason for the insolubility and unexplained elemental analysis results of MS products. The presence of the Fe impurities prohibits the use of solid-state NMR. Hence, XPS was used to investigate bonding information of the MS polymers by recording the C 1s, S 2p, Fe 2p and O 1s regions (Fig. 2C). As Fig. S13 shows, clearly there are more S and C chemical environments observed from MS(S-DIB) or MS(S-Styrene) than that of TS(S-DIB) and TS(S-Styrene). Fig. 2F clearly shows the presence of Fe in the polymer MS(S-Styrene), and Figs. 2D and 2E give the detailed connectivity information of polymer MS(S-Styrene). XPS S 2p peak in Fig. 2D is fitted by three components: neutral S (163-165.2 eV) and cationic S+ (165.3-166.7 eV) and oxidized SO3− (166.8-168.4 eV)36. The S 2p spectrum was curve fitted, indicating the presence of four sulfur chemical bonding environments; S-S, C-S, S-O and S with Fe at a binding energy (BE) of 163.8 eV, 165.0 eV, 165.4 eV/166.6 eV and 166.9 eV /168.1 eV, respectively5, 37, 38. Furthermore, the bonding assignments from the C 1s data, Fig. 2E, show five carbon bonding environments; C-C, C-S, C-O, C=O and metal carbonate at a BE of 284.7 eV, 285.5 eV, 286.4 eV, 287.9 eV, 289.3 eV respectively.36, 39 That means that sulfur has reacted with the organic co-monomers and chemically connected with carbon to form C-S bonds, and there are bonds of metal sulfonate and metal carbonate formed in the meantime. In contrast, XPS S 2p and C 1s regions (and accompanying survey scan) of TS(S-Styrene) (Fig. S13d) show no Fe present, carbon and sulfur were only observed in only the following bonding environments; S-S (163.9 eV), C-S (165.0 eV in S 2p spectrum and 285.49 eV in C 1s spectrum), and C-C (284.7 eV in C 1s spectrum). Hence, it is further demonstrated that new covalent bonds of C-S were formed in MS polymer, and Fe not only disperses in the polymer physically but chemically connects with the polymer chains. Moreover, XPS spectra of MS(S-DIB) and TS(S-DIB) show similar results with that of MS(S-Styrene) and TS(S-Styrene) (Figs. S13a and S13b). Therefore, all the above proof illustrates that sulfur did react with C=C bonds in the monomers initiated by mechanical energy to successfully form the sulfur polymers, and iron from the steel balls reacted with the polymer to form some inorganic proportion accompanying the polymer formation rather than only being ground iron dispersed among polymer particles, which is surprising and interesting.
Solution inductively coupled plasma optical emission spectrometry (ICP-OES) was used to analyse Fe content of the polymers. As Table S3 shows, there are ranging from 8 wt.% to 20 wt.% Fe in the polymers but there is only 0.77 wt.% Fe in the control sample--ball milled pure elemental sulfur. Furthermore, thermogravimetric analysis (TGA) of MS polymers in N2 shown in Fig. S14 illustrates that there still are some residue percent from polymers at 1000°C, but only around 1.4% residue of control sample (ball milled sulfur) was obtained. It is revealed that Fe only reacts with polymer chains during polymer formation but does not react with pure sulfur. Now, we can understand that why the ratios of C:H and C:S decrease after polymerization in EA results. That is because that percentage of metal cannot be detected using EA technic, but the total percentages of elements were calculated basing on total polymers’ mass, which causes decrease of carbon percentage. In addition, unreacted monomers were washed out by acetone before the EA characterization, which further decreases the carbon percentage. Hence, the detected carbon percent is lower than the calculated value so that the ratios of C:H and C:S are lower than the calculated values. That also indicates that there may be no/less H2S formation during reaction as there is no decrease of sulfur percentage. Theoretically, the organic material all can be burned off in the air. However, the synthesized materials in this case still have Fe oxide remaining after being burned, which can be proved by the EDS images of burned MS(S-DIB) in Fig. S15, there is only elements Fe, Ge or some C which cannot be confirmed since the background is carbon film, but no sulfur remaining. Hence, it is clear that an inorganic fraction formed and iron from the steel balls cause the questions discussed above. Fe reacted with the polymer chain and causes structure change to the polymer resulting in insolubility, where Fe acts as a kind of crosslinker to link the polymer chain into the network. While, the FT-IR curves of some of the polymers in Figs. S16-20 show that partial reaction of C=C of monomer containing 2 or more vinyl groups causes those MS polymers to have lower Tg than the TS polymers. Whereas, some monomers which potentially form highly linear structured polymer, like styrene which has the least C=C so that it has high sulfur rank and high opportunity for Fe to attend the reaction to form high content of inorganic fractions, resulted in the formed polymer having higher Tg than the TS polymer.
In addition, H2S gas measurements were done to evaluate the hydrogen adsorption in both of MS and TS polymerization. As it is hard to monitor the reaction during the reaction due to the practical limitations of the equipment, the products were used to analyse H2S generation tests. Polymers TS(S-DIB), TS(S-DCPD), TS(S-DVB) and TS(S-ENB) were chosen as the control samples to compare with the MS polymers from the same crosslinker respectively, as they all are solid state at room temperature. From Fig. 3A, it can be seen that H2S is only generated under heating. Slight H2S concentration can be detected from some of MS products once the temperature reaches 80°C, but a significant H2S concentration can be detected when the temperature is 140°C no matter whether MS polymers or TS polymers. MS products show quicker H2S generation rate than TS products as they are smaller particles with higher surface area and shorter diffusion pathways. Even through the TS polymers are fully crosslinked polymers, there was significant H2S generation at high temperature. This suggests that H2S gas is likely released during the TS process thanks to the high reaction temperature (normally higher than 140°C). Instead, it can be considered as that there is likely no/less H2S gas generated during the MS process due to absence of heating. Additionally, the H2S concentrations released from MS polymers are all higher than that from TS polymers (the data for every polymer can be found in Figs. S24-S31). In theory, less gas generated from the products suggests hydrogen abstraction may have already occurred during the synthesis process as the same monomers and ratio were used in synthesis. Therefore, for the reaction itself, there should be less or no H2S generated during the MS process, in comparison to the TS process. Moreover, scanning electron microscopy (SEM) images of the MS polymers (Figs. S32-S39) show all the polymers are relatively small size particles. Accordingly, the obtained polymer powders were considered as a sorbent in water remediation. Hence, mercury (Hg) uptake tests were carried out using all the MS polymers with the control samples of solid-state TS polymers and pure sulfur. The experimental procedures of Hg uptake can be found in the SI. As Figs. 3B show, all the MS polymers have higher Hg capture efficiency than the TS polymers as well as pure sulfur. The highest ratio 62% Hg was removed from 138 ppm HgCl2 solution by the polymer MS(S-Myrcene) in 24 hours and the highest capacity of 42.3 mg/g was obtained, which is higher than normally non porous sulfur polymers and even higher than some kinds of porous polymers reported22, 40, 41, such as salt templated sulfur polymer which has the capacity of 2.27 mg/g.40 Additionally, the MS polymer powders can be processed furtherly by hot pressing into polymer thin film, as shown in Fig. 3C, despite their crosslinked structures. As the polymers contain disulfide bonds, the polymer thin film was expected to have potential for healing. It is known that dynamic reaction between the disulfide bonds can be introduced by heat42 and also ultraviolet (UV) light43. Thermally-induced healing properties of inverse vulcanized polymers has been widely investigated8, 9, but we demonstrate here first time that the inverse vulcanized polymer is able to be self-healed under UV light irradiation. Fig. 3D indicates that two pieces of MS(S-ENB) films were healed together after radiation using 285 nm UV light for 40 minutes. The healed film shows a good elastic property as the video S1 shows, where the film can be stretched and then the deformation can be recovered automatically once the force is released.
{kind=link}