This study sought to overcome the difficulties associated with using small amounts of exosomal mRNA in breast cancer research.
According to the WHO, cancer is a leading cause of death worldwide, with breast cancer being the fifth most common cancer and one of the most common malignancies in women.(16) Breast cancer is a heterogeneous disease with several sub diseases with differences in tumor biology, histopathology, and prognosis. About 20% of all breast cancers are characterized by amplification of 17q12, leading to overexpression of epidermal growth factor receptor 2 (ERBB2/HER2/neu).(17)
ERBB2 is a transmembrane tyrosine kinase with oncogenic potential and belongs to the epidermal growth factor (EGF) receptor family. Since identification of ERBB2 amplification in breast cancer, multiple genes have been reported to be co-amplified with ERBB2 and a 2 Mb region at the 17q12–21 and their relation to trastuzumab (Herceptin) resistance.(18) The 17q12-q21 region has been heavily implicated in breast cancer and was identified as a common region of amplification and overexpression in breast tumors.(19–21) In this study, we observed the expression levels of ERBB2, along with three genes (PPP1R1B, GRB7, and STARD3) that are commonly co-amplified with ERBB2, in breast cancers.
We designed a mouse model using breast cancer cell line HCC1954, in which the ERBB2 gene is overexpressed, to determine the degree of expression of ERBB2 co-amplified genes (Table 1).(22–26) One of the several recently developed systems to enrich exosomes efficiently from culture supernatant and liquid biopsy was used in our study.(27)
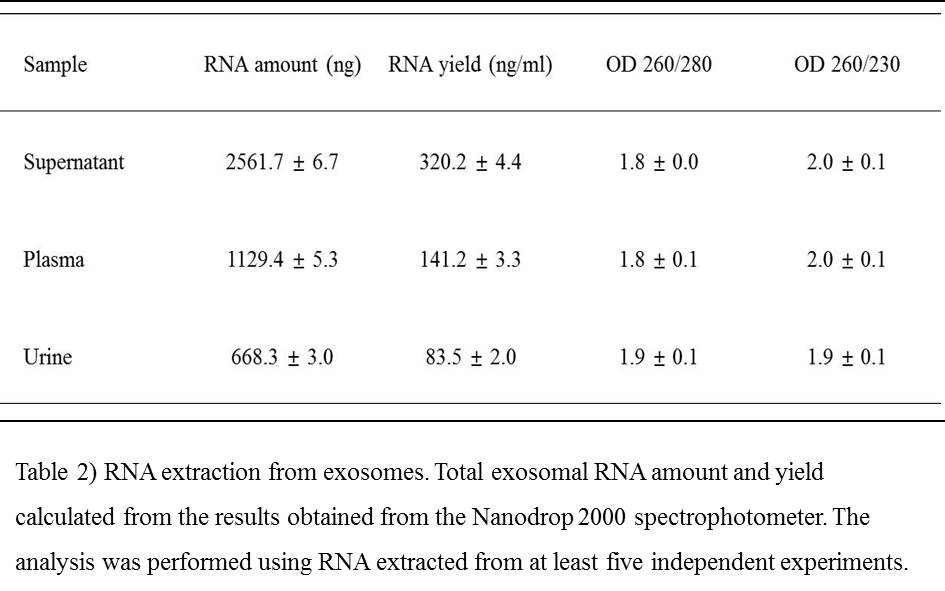
Electron microscopy confirmed that the structures studied were exosomes (Fig. 1 A). We determined the quality of the RNA collected from supernatant, urine, and plasma using the Agilent 4200 Tapestation system. All samples yielded RNA of equivalent size distribution and abundance (Fig. 1 B), indicating good-quality RNA including 18S and 28S rRNA peaks. In contrast, exosomal RNA differed in profile due to absence of two peaks. Since the algorithm is based on ribosomal RNA, little of which is contained in exosomes, RNA integrity number (RIN) values are only valid for cellular RNA quality assessments.(28–30) For this reason, RNA purity was evaluated spectrophotometrically at absorbances of 230, 260, and 280 nm (Table 2). Obtaining high-quality RNA is a critical step in many molecular techniques, such as RT-qPCR and transcriptome analysis by next-generation sequencing or hybridization to microarrays. The quality and reproducibility of RNA sample preparation is crucial for diagnostic development, especially for clinical samples.(31)
Exosome isolation has limitations including difficult extraction of exosomal RNA from urine and plasma in mouse models. To overcome these shortcomings, we used a two-stage PCR method. To reduce PCR errors caused by primer dimers and/or nonspecific fragments, which may occur in multiplex-PCR amplification, we used Pfu-DNA polymerase with lower error rate than taq polymerase and limited the pre-amplification step of the first PCR to 13 cycles.(32, 33)
We compared the conventional PCR protocol with a two-stage PCR protocol using the same primers and cycle and amplification conditions. The quality values of the amplicons produced by the two-stage and conventional PCR were compared using the Sanger method. Sequencing of the two-stage PCR-derived products revealed no changes in product sequence compared to that of the conventional method, and the obtained sequences were queried using BLAST, which revealed a 98–99% match to the target gene sequences in the database (Fig. 2 B).
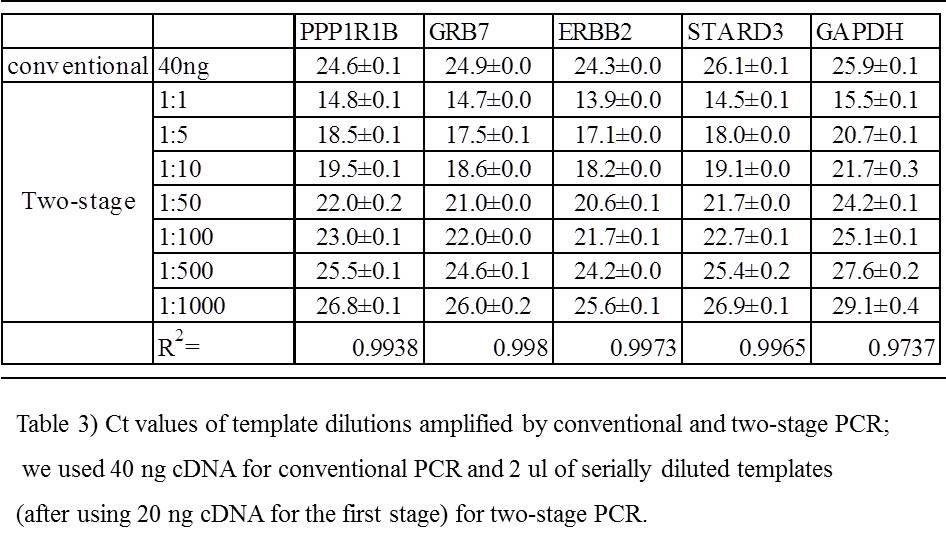
We compared the amplification cycle threshold (Ct) values of four target genes from the conventional PCR and the two-stage PCR templates serial dilution, and found similar Ct volumes of two-stage PCR template dilution (1/200).
Conventional PCR detected some sample errors introduced during amplification, detected from meting curve analysis. This suggests the presence of primer dimers or nonspecific fragments.(34) On the contrary, the two-stage PCR templates showed stable melting curves, is same results at serial dilution templates (Fig. 3).
In quantitative RNA expression analysis of all samples by conventional PCR compared to those of two-stage PCR template dilution (1/200), similar Ct volumes were observed for all sample groups (cell, supernatant, urine, and plasma) and same result of cell and supernatant groups. But it showed different aspect of urine and plasma groups in relative quantitation (Fig. 4). While the expression of target gene seems to have decreased in the conventional PCR, the expression of target gene appears to be increasing in the two-stage PCR, especially in the plasma group.
{kind=link}
{kind=link}
{kind=link}