Tumors trend to adapt to the changes of microenvironments when they are threatened by death. In clinic, some tumors always stay in quiescent condition because of the hypoplasia of their blood vessels. Some tumor tissues remain dystrophic since they can not obtain enough nutrient substances through hypoplastic blood vessels. What’s more, selectively starving cancer cells can also make tumors keep in malnutrition, which is the metabolic-based therapy for cancers with tiny side effects. Cancer-starving therapies, such as dietary modification, tumor angiogenesis inhibition and aspartic acid deficiency, can effectively decrease the incidence of spontaneous tumors and slow the growth of primary tumors[14].
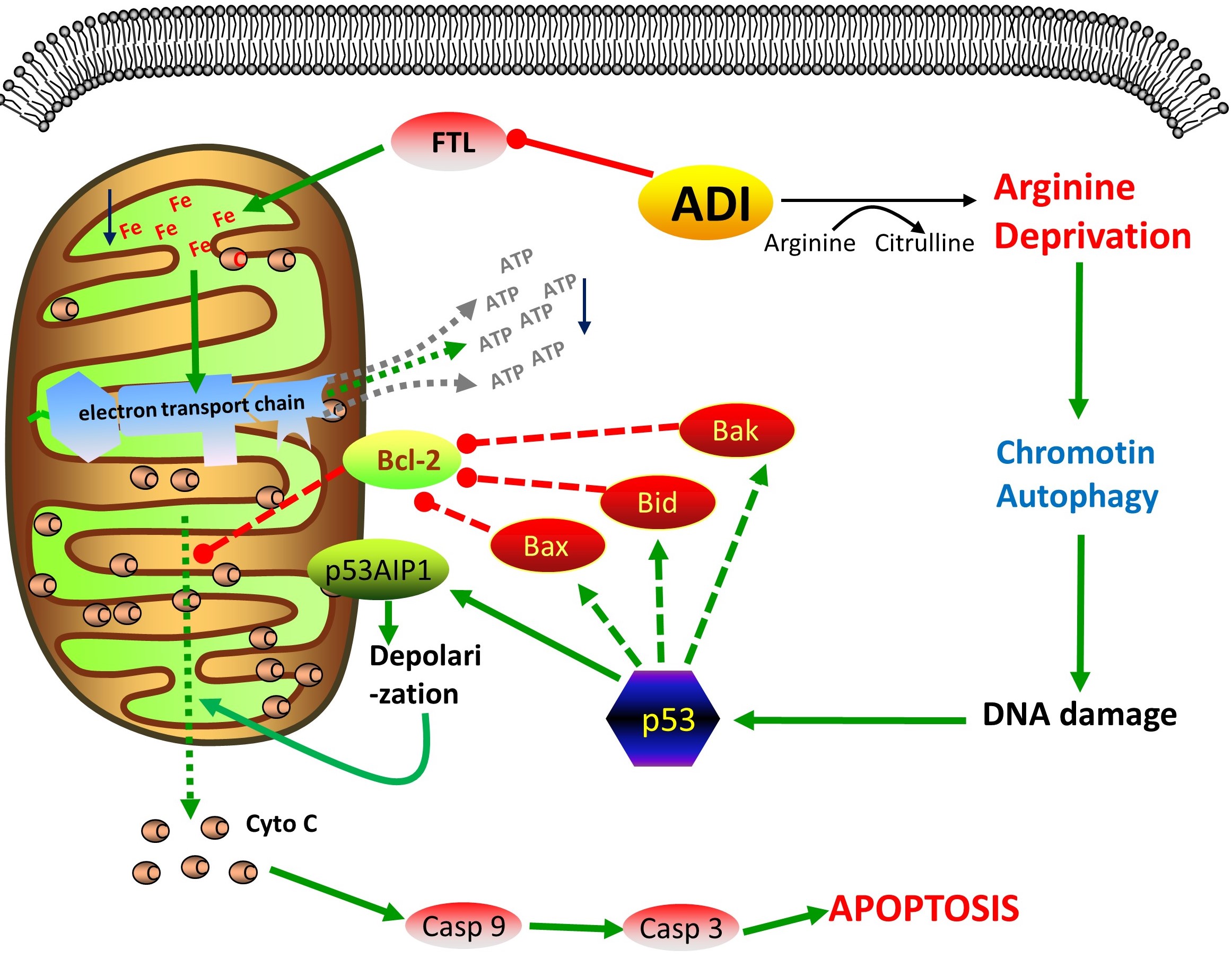
ADI was a good gene with the potential for cancer gene therapy. As the description of our preliminary work[9], cytosolic ADI displayed higher apoptosis-inducing efficiency, tumor-targeting specificity, and oncolytic activity[9]. In order to exclude the actions of adenovirus on cells, we just used pcDNATM4/TO/myc-His vector as ADI expression vector without replacing pCMV promoter with phTERT promoter. Rapid growth of tumors requires a lot of nutrition including arginine. Tumor cells with ASS gene deficiency are more sensitive to arginine deprivation than normal cells, for example endometrial cancer[15]. Based on cancer tissue specificity of ASS expression[4], we used MRC5 (ASS+), PC3 (ASS-) and HepG2 (ASS-) cell lines to explore whether ADI had the same effect on different cancer cell lines. As shown in fig 1, ADI expressed in cytosol eventually induced cellular apoptosis of PC3 and HepG2 cells.
ADI-PEG20 had been proved to induce cellular autophagy and caspase-independent apoptosis through exhausting the arginine in peripheral microenvironment of tumor [16]. However, it is unknown whether ADI expressed in cytosol has the same anti-tumor mechanism. We expect to understand whether ADI has the unique anti-tumor mechanism in vivo. Therefore, we screened the protein factors that would interact with ADI by yeast hybrid method. FTL was screened out as shown in fig 2. Co-IP results proved the interaction of ADI and FTL in cells. Fluorescence co-localization displayed that the interaction happened in the cytoplasm.
Ferritin is considered the major iron storage protein, which participates the regulation of cellular iron homeostasis[17]. Mitochondrial function also requires iron replenishment from cytoplasmic ferritin. The inhibition of ferritin directly results in the dysfunction of mitochondrial electron transport[18]. In order to exclude the effect of ADI enzyme activity on cell function, the catalytic residues of ADI were mutated into alanine residues. Cysteine398, the catalytic residue of ADI[10], was mutated into alanine398. Since alamine398 as an inert residue has no nucleophilic catalytic capacity, the mutation (C398△A398) effectively terminated the enzyme activity of ADI[19]. As shown in fig 3, ADI△(C398△A398) still caused a small number of cell death in PC3 and HepG2 cells. The overexpression of FTL neutralized the death-induced effects on these two cells. We speculated that FTL overexpression made up the part of cytosolic FTL that has lost its function due to interaction with ADI. However, ADI△(C398△A398) need 3 days to induce cancer cell death, but ADI only need 2 days as shown in fig 2. Obviously, cytosolic ADI△ just induces limited apoptosis through interacting with cytosolic FTL. The interaction of ADI and FTL is not the main reason of mitochondrial damage. In addition, as shown in Fig. 1, the high concentration of arginine in the culture medium counteracted the cell death caused by cytosolic ADI. This result further demonstrates that arginine deprivation in cytosol is the main mechanism for cytosolic ADI to suppress cancer cells.
Accumulating evidences in research papers have illustrated that arginine deprivation in vitro exerts its anticancer effects in various tumors by inducing mitochondrial damage and autophagy[5, 6, 20, 21]. Arginine deprivation inhibits nitric oxide synthesis in cells[22, 23]. Thus, arginine deprivation can not damage mitochondria by increasing nitric oxide biosynthesis in cells. David K. Ann and Hsing-Jien Kung[24] also reported that mitochondrial damage is the major reason of cancer cell apoptosis induced by ADI-PEG20. Our MPTP experiments also proved that cytosolic ADI led to serious mitochondrial damage as shown in fig 4c. However, it is still unknown for the apoptosis pathway induced by mitochondrial damage during arginine deprivation in vivo.
Then we checked the expression of some protein factors relating with mitochondrial apoptosis pathway. As shown in fig 4a and 4b, 2 days of arginine deprivation in vivo increased the expression of p53 and p53AIP1 proteins in PC3 and HepG2 cells. The ectopic expression of p53AIP1 protein will induce the down-regulation of mitochondrial Δψm (transmembrane potential) and the release of cytochrome c from mitochondrial by interacting and inhibiting Bcl-2 at mitochondrial[25]. Obviously, after two days starvation, the increased expression of p53AIP1 protein activated the p53-dependent apoptosis by interacting with the same upregulated expression of p53 protein[11, 26]. Then cytochrome C was released from mitochondrial. Casp3 and Casp9 were activated as shown in fig 4d and 4e. At the later stage of arginine deprivation in cells (for 4 days), PC3 and HepG2 cells seemed to enter the initiative apoptosis process, because the increasing expression of Noxa, PUMA, Bax and Bak proteins would further aggravate mitochondrial damage[27, 28] as shown in fig 4a and 4b.
We further knocked down the mRNA levels of p53 and p53AIP1 to verify their action during arginine deprivation in cells. As shown in fig 5a, 5b, 5d and supplementary fig S5, the knockdown effectively reduced the apoptosis rates in PC3 and HepG2 cells. p53 knockdown displayed the better effects on apoptosis inhibition than p53AIP1 knockdown. Mitochondrial damage was also prevented by p53 knockdown, due to the higher fluorescence intensity of living cells exhibiting in fig 5c. Therefore, p53-dependent apoptosis pathway was the major pathway induced by cytosolic ADI.
Mitochondrial damage is not the only way to result in cancer cell death during arginine deprivation in cytosol. Cell autophagy was reported to be induced by ADI-PEG20[16]. Autophagy, the process of cellular self-eating, is usually caused by starvation or stress, which is capable to degrade long-lived proteins and organelles such as endoplasmic reticulum, mitochondria, peroxisomes, ribosomes and nucleus[29, 30]. We also verified that autophagy was induced by cytosolic ADI. pEGFP-LC3 and pcDNA4-ADI plasmids were co-transfected into cells. With the expression of ADI, the more proteins were converted from LC3-I to LC3-II as shown in figure 6b. Cytosolic GFP-LC3-I will be conjugated to phosphatidylethanolamine to form GFP-LC3-II during autophagy. GFP-LC3-II will be recruited into autophagosomal membranes with the formation of autophagosomes[31]. As shown in figure 6a, green GFP fluorescent particles presenting around the nucleus were autophagosomes in two cancer cells. Thus, with the expression of ADI, the autophagy induced by arginine starvation was indeed taking place in the cells.
Hsing-Jien Kung reported that arginine deprivation in vitro will lead to cancer cell chromatin autophagy[32]. He indicated that prolonged arginine deprivation causes mitochondria dysfunction and ROS generation, eventually resulting in nuclear DNA damage and membrane remodeling. Excessive autophagy leads to giant aggregate of autophagosomes/autolysosomes fusion at the later time point of arginine deprivation in vitro. Stephen Gregory[33] disclosed that chromatophagy is necessary for the survival of chromosomal instability (CIN) cells. Chromatophagy is activated to remove the defective mitochondria in response to DNA damage. However, we had an additional view about the chromatophagy. We think that arginine deprivation will mobilize cells to utility endogenous arginine storage. Nucleosome, especially histone 3 (H3), contains abundant arginine residues. Therefore, cells will have to obtain arginine from chromatophagy to maintain basic physiology during arginine deprivation. As shown in fig 6f, nucleus budding presented in HepG2 and PC3 cells after the co-transfection of pcDNA4-ADI and pEGFP-LC3 for 96 hours. Chromatin fragment (blue fluorescence) and H3 proteins (red fluorescence) were displayed to co-localize in autophagosomes (GFP green fluoresence). This showed that ADI expressed in cytosol also induced chromatin autophagy. H3 proteins presenting in autophagosomes implied the utility of histones arginine.
{kind=link}