SRGN is an Independent Prognostic Factor and Potential Serum Biomarker in HCC Patients
To investigate the role of SRGN in HCC progression, SRGN mRNA expression in primary tissues was analyzed in the GSE364 cohort. SRGN was upregulated in metastatic primary tissues compared with that in non-metastatic primary tissues (Fig. 1A). Additionally, we found that SRGN expression was significantly higher in tissues with metastasis than in those without metastasis in TCGA database (Fig. 1B), a finding that was similar to that in the GSE364 cohort. Moreover, SRGN expression was significantly higher in lymph node metastasis tissues than in non-lymph node metastasis tissues (Fig. 1C). Serum tumor markers were commonly used as a diagnostic method for HCC, and we speculated that SRGN could be another significant biomarker for HCC diagnosis. To determine the significant role of SRGN in HCC progression, we collected 123 serum samples to evaluate the clinical implications of SRGN expression.
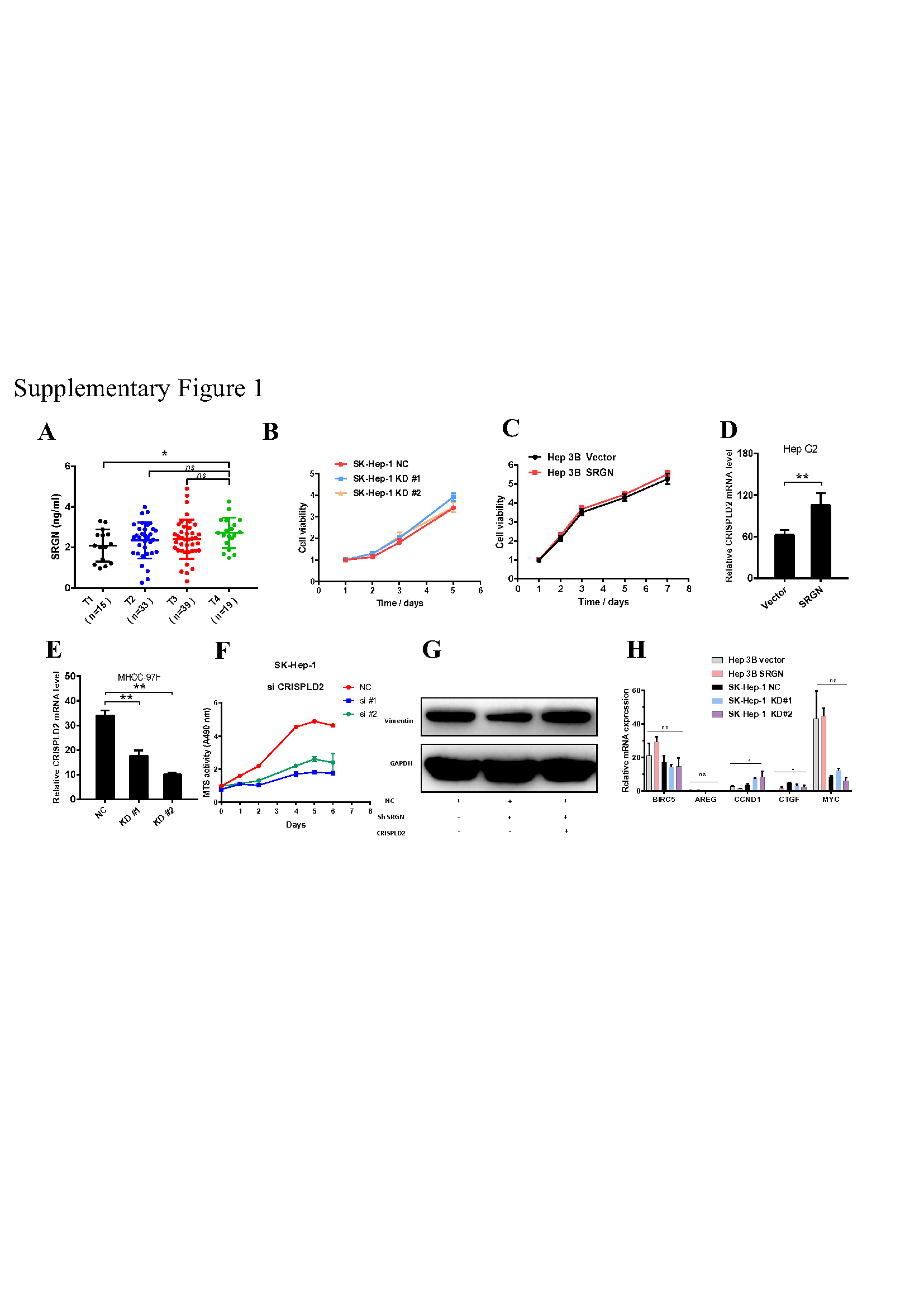
To confirm this hypothesis, we performed ELISA and examined SRGN protein expression, which was significantly higher in the serum of patients with HBV than in the serum of patients without HBV (Fig. 1E). The level of SRGN protein in liver cancer patients without metastasis was higher than that in healthy volunteers, and even higher in patients with metastasis (Fig. 1F). SRGN in the serum of HCC patients was positively correlated with diameter of neoplasms (Fig. 1G), AFP (Fig. 1H), clinical stage (Fig. 1I) and T stage (Supplementary Fig. 1A). Therefore, we elucidated that SRGN is a potential serum biomarker and plays a critical role in HCC progression.
To evaluate the prognostic value of SRGN expression in primary HCC tissues, we searched the TCGA database of SRGN expression in HCC samples. High expression of SRGN showed a significant overall survival outcome when the survival time was more than 700 days (Fig. 1D). In our collected serum samples, we found that high expression of SRGN was showed a significant overall survival (OS) outcome (Fig. 1J). Multivariate analyses further indicated that elevated SRGN expression is an independent, unfavorable prognostic indicator in HCC patients (Fig. 1K). Taken together, these analyses revealed that high SRGN levels in HCC are significantly correlated with poor patient outcomes.
The proteoglycan SRGN Promotes HCC Cell Migration and Metastasis in an ECM Autocrine Manner
To confirm the findings described above, quantitative real-time PCR and immunoblotting were performed to quantify the mRNA levels of SRGN among the HCC cell lines, SRGN mRNA and protein expression were differentially expressed among the different HCC cell lines (Fig. 2A). To explore the role of SRGN in HCC cell growth, we transfected SK-Hep-1 cells with shRNA or negative control. Notably, we confirmed that secreted SRGN protein of approximately 300 kDa in molecular weight was significantly decreased in the conditioned medium (CM) (Fig. 2B and 2C). We observed the SRGN suppression was not significant in HCC proliferation ability (Supplementary Fig. 1B). Additionally, we generated cell lines overexpressing SRGN in Hep 3B cells, and SRGN protein expression was validated by qPCR and immunoblotting (Fig. 2B and 2C). Overexpression of SRGN in Hep 3B showed a similar result, with no significant difference in the HCC proliferation ability (Supplementary Fig. 1C).
To explore the relationship between SRGN and the metastatic process, we performed wound healing assays to evaluate the migratory ability of SRGN. SRGN knockdown dramatically reduced the migratory ability, and SRGN overexpression increased the migratory ability (Fig. 2D and 2E). To confirm this result, we performed the transwell assay, which showed that SRGN knockdown largely reduced the migratory ability, and SRGN overexpression promoted migration (Fig. 2F, G). These results indicate that SRGN specifically functions by promoting migration in HCC cell lines. However, we found that the knockdown of SRGN expression resulted in the downregulation of the levels of EMT-promoting factors, such as N-cadherin, vimentin, and MMP2, and the upregulation of epithelial markers, such as E-cadherin, in SK-Hep-1 cells (Fig. 2H). Conversely, SRGN overexpression resulted in the upregulation of N-cadherin, vimentin and MMP2 and downregulation of E-cadherin in Hep 3B cells.
To validate the metastatic function of SRGN in HCC cell lines in vivo, SRGN KD #2 and control cells were injected into the tail veins of nude mice. After 8 weeks, the mice were euthanized and their lungs were excised and examined. Using hematoxylin and eosin (H&E) staining, the metastatic nodules in the lung tissue of SRGN KD#2 mice were observed to be fewer and smaller than those in the control mice (Fig. 2I and 2K). The bodyweight of the KD #2 group was higher than that of the control group (Fig. 2J). These results strongly suggest that SRGN is closely associated with metastasis in HCC cells.
SRGN is Significantly Associated with Stemness-Like Properties in HCC Cells
Recent studies have shown that CSCs are highly metastatic and may be quite plastic and associated with EMT. To explore whether SRGN can impact cancer stem cell ability, we used MHCC-97H cells, a suitable cell line model to perform the self-renewal assay, and the knockdown efficiency is shown in Fig. 3A and 3B. To examine the effect of SRGN on maintaining cancer stem cell characteristics, we performed a sphere culture assay, and SRGN knockdown reduced the number of spheres generated by MHCC-97H cells (Fig. 3C and 3D), indicating the critical role of SRGN in the self-renewal capacity of the cells. CSCs have the capacity for self-renewal and differentiation potential, and previous studies [26] have demonstrated that the side population (SP) cells, identified by their ability to pump out a fluorescent dye (Hoechst 33342), have certain characteristics of CSCs, the SP phenotype can be a CSC marker for HCC. Herein, we found that the side population in MHCC-97H cells with SRGN knockdown was reduced compared with control cells (Fig. 3E and 3F). Overexpression of Hep 3B cells with SRGN showed that ABCG2, Bmi-1, and Nanog were largely increased. Additionally, we suppressed SRGN in MHCC-97H cells and tested the three different CSC markers. ABCG2, Bmi-1, and Nanog were reduced at the protein level, indicating a promoted HCC self-renewal capacity (Fig. 3G). More importantly, the SP sorting assay revealed that SRGN was upregulated in the side population, indicating more CSC-like ability in HCC (Fig. 3H). These observations suggest that SRGN is closely associated with the CSC-like properties of HCC cells.
SRGN with its Receptor CD44 Regulates the Nuclear Localization of the Hippo Pathway Effector YAP
Our previous study[14] showed that SRGN induces CD44 expression to potentiate its self-renewal capacity by activating the MAPK pathway in NPC, and CD44 was a receptor of the ECM ligand SRGN. Therefore, we examined the expression of CD44 and SRGN in HCC. The relative expression level of CD44 was increased and decreased after SRGN suppression and overexpression, respectively (Fig. 4A). Notably, the TCGA database showed a positive correlation between CD44 and SRGN in primary HCC tissues (Fig. 4B). These results indicate that SRGN expression is positively correlated with CD44 expression in both HCC cell lines and tissues.
The Hippo/YAP pathway has been delineated and shown to play an important role in regulating tissue proliferation and apoptosis rates. It is highly expressed in stem cells and associated with the CSC capacity. Based on the result that SRGN can significantly promote self-renewal ability, we hypothesized that promotion of CSC ability by SRGN might be associated with the Hippo/YAP pathway.
To further explore whether SRGN plays a crucial role in the YAP pathway, we found that SRGN was positively correlated with YAP expression in HCC cell lines (Fig. 4C). In mammals, MST1 is a core kinase cascade in the YAP pathway that can phosphorylate the kinase LATS1, and then the LATS1 complex phosphorylates and represses the transcriptional coactivator YAP. We also found that the phosphorylation of YAP was decreased and the phosphorylation of MST1 and LATS1 was increased, as detected by immunoblotting (Fig. 4D). Thus, SRGN suppresses the YAP pathway via inhibiting YAP phosphorylation. When SRGN is overexpressed, YAP expression was increased, particularly in the nucleus (Fig. 4E). To confirm this result, we performed the confocal immunofluorescence assay to examine the nuclear and cytosolic/membrane fractions and found that SK-Hep-1 knockdown cells with less YAP translocated to the nucleus (Fig. 4F), whereas more YAP translocated to the nucleus in SRGN-overexpressed Hep 3B cells (Fig. 4G). The analysis of SRGN knockdown and overexpression cells revealed that SRGN affects YAP expression in the nucleus significantly. To elucidate the function of SRGN in HCC progression, we generated the DEN/CCL4 induced-HCC mouse model to evaluate the correlation of SRGN and YAP in the process of HCC mouse model formation. Confocal immunofluorescence assay showed that HCC tumors formed in the mouse model, increased YAP and SRGN expression was observed in liver tumor tissues, and YAP was highly expressed in the nucleus (Fig. 4H). Thus, SRGN regulates YAP expressionand and the nuclear localization of YAP in HCC cells and mouse models.
CRISPLD2 is a SRGN-Mediated HIPPO/YAP Pathway Downstream Effector Gene and Potentiates HCC Cell Motility Ability
To explore the downstream targets of SRGN that promotes HCC metastasis, we performed whole-genome expression profiling in SK-Hep-1 cells and Hep 3B cells. Given that SRGN promotes cancer metastasis, we focused on the SRGN-regulated genes implicated in these processes from microarray analysis (Fig. 5A). Notably, CRISPLD2, also named LGL1, overlapped in the knockdown and overexpression groups, and the increased expression of CRISPLD2 by SRGN overexpression was validated by qPCR and immunoblotting at the mRNA and protein levels, respectively (Fig. 5C,D; Supplementary Fig. 1D, E), and SRGN was highly correlated with CRISPLD2 in HCC cell lines and TCGA database (Fig. 5B, C). Using siRNAs to suppress CRISPLD2, we found that vimentin expression was also decreased (Fig. 5G), a finding that was similar to the result after the knockdown of SRGN expression. Cell growth assays showed that the knockdown of CRISPLD2 can significantly inhibit cell proliferation (Supplementary Fig. 1F). The Transwell assay showed that CRISPLD2 knockdown can inhibit cell migration ability (Fig. 5E). Overexpression of SRGN in Hep 3B cells was validated by qPCR and immunoblotting (Fig. 5F), and the migration assay showed that SRGN promotes metastasis in vitro (Fig. 5E).
The YAP-TEAD1 protein-protein interaction (PPI) mediates the oncogenic function of YAP, and inhibitors of this PPI have potential use in the treatment of YAP-involved cancers [27]. Peptide 17 is a promising inhibitor that can efficiently disrupt the interaction between YAP and TEAD1. Peptide 17 induction can inhibit the SRGN overexpression effects in increasing CRISPLD2 expression, while with no effects observed in the vector group (Fig. 5H). Immunoblotting showed that the CRISPLD2 protein level decreased after peptide 17 induction in the SRGN overexpression group compared with that in the vector group (Fig. 5I). Thus, peptide 17 can suppress CRISPLD2 expression by inhibiting SRGN expression.
To further confirm that ECM proteoglycan ligand SRGN induces the expression of its target gene CRISPLD2, CRISPLD2 promoter luciferase reporter assay revealed increased activity with SRGN overexpression. However, after peptide 17 induction, the regulation of CRISPLD2 activity by SRGN was suppressed (Fig. 5J, K). The transwell assay showed that suppressed SRGN could decrease the migration ability. Additionally, CRISPLD2 overexpression after SRGN suppression increased the migration ability to some extent (Fig. 5L, M, N; Supplementary Fig. 1G).
Next, we determined whether SRGN promotes the migration ability in the YAP pathway. We treated the cells with verteporfin (VP), which disrupts the formation of the YAP-TEAD1 complex, significantly reducing preneoplastic foci and oval cell proliferation. Treatment with different doses of VP decreased the migration ability of SRGN-overexpressing cells. CRISPLD2 suppression reversed the SRGN-promoting migration ability (Fig. 5O). Collectively, these results strongly suggest that the promotion of HCC metastasis by upregulating SRGN depends on the up-regulation of CRISPLD2. CRISPLD2 is a SRGN-mediated HIPPO/YAP pathway downstream effector gene and potentiates HCC cell motility ability.
CRISPLD2 Is a Novel YAP-TEAD1 Target Gene Dependent on SRGN
To further confirm that SRGN mediates the downstream target gene CRISPLD2 and YAP pathway, we transiently transfected MHCC-97H cells with YAP. As expected, the CRISPLD2 promoter luciferase reporter assay revealed increased activity of the CRISPLD2 promoter with YAP overexpression (Fig. 6A). When treated with verteporfin, the CRISPLD2 promoter was suppressed (Fig. 6B), suggesting that the activity of the CRISPLD2 promoter was directly regulated by the YAP-activating specific signaling pathway.
We used the Jasper database (http://jaspar.binf.ku.dk/) to predict that the co-transcription factor YAP-TEAD1 has two potential binding sites in the promoter region of CRISPLD2. The luciferase assay proved that the deletion of site 1 reduced the activity of luciferase, but the deletion of site 2 showed no significant change in the activity of luciferase (Fig. 6C). Thus, the co-transcription factor YAP-TEAD1 accelerates CRISPLD2 transcription through binding site 1 (TGGATTCCTGGG).
To confirm the mechanism study, we used chromatin immunoprecipitation (ChIP). Based on the CRISPLD2 promoter region, we designed 5 pairs of PCR primers (Fig. 6D). The CRISPLD2 promoter sequence was detected by qPCR and normalized to the input level (Fig. 6E and 6F).The data revealed that YAP-TEAD1 directly binds to the CRISPLD2 promoter and activates CRISPLD2 transcription, and two binding sites in CRISPLD2 promoter are regulated by YAP-TEAD1. However, YAP/TEAD1 target genes (BIRC5, AREG, CCND1, CTGF and MYC) were not activated by SRGN-mediated HIPPO/YAP signals in HCC cells (Supplementary Fig. 1H), indicating that CRISPLD2 is a novel selective YAP-TEAD1 target gene in the SRGN-mediated HIPPO/YAP signaling pathway.
Cotreatment with the YAP-TEAD1 inhibitor and sorafenib inhibits tumor growth in vivo and in vitro
We further investigated whether SRGN directly promotes the sorafenib resistance by performing flow cytometry and measuring the percentage of apoptosis 2 days after sorafenib induction. In the KD#1 and KD#2 groups, as the drug concentration increased, the apoptosis rate was increased less than that in the control group (Fig. 7A, C). Additionally, sorafenib largely induced apoptosis in SRGN overexpression cells than vector cells in a dose-dependent manner (Fig. 7B, D).
To further explore the signaling pathways induced by SRGN under sorafenib treatment, we first checked the total phosphorylated ERK, P-ERK, YAP and P-YAP in SK-Hep-1 and Hep 3B cell lines. Hep-3B cells treated with sorafenib after overexpressing SRGN, the hallmarks of apoptosis such as the induction of caspase-3 cleavage occurred in a dose-dependent manner (Fig. 7E, F), and the effect of sorafenib on the YAP signaling pathway in HCC cells indicated that targeting SRGN can inhibit cell apoptosis in the YAP pathway. Sorafenib could abrogate the stimulatory effects of SRGN on YAP, P-YAP, ERK, and P-ERK. Moreover, we found that overexpressing cells had higher cleaved caspase-3 levels than vector cells, but they had lower caspase-3 levels than the vector cells in a sorafenib dose-dependent manner.
The cell viability assay showed that VP could reverse the suppression of sorafenib-induced cell apoptosis by SRGN-overexpressing cells (Fig. 7G and 7H). Additionally, colony formation showed that VP and sorafenib combination treatment could largely decrease the cell colony formation ability (Fig. 7I and 7J). The confocal immunofluorescence assay showed that YAP was highly expressed in nuclei of the side population, revealing that YAP likely translocates the nucleus compared with the main population (Fig. 7K).
The SP test showed that verteporfin could significantly reduce the side population from 14.67–0.29%, while sorafenib can reduce the cells of the main population from 10 × 106 to 6.87 × 106 but not those in the verteporfin group (Fig. 7L, M). Sorafenib combined with verteporfin greatly reduced the main and side populations of MHCC 97H cells.
Next, an SK-Hep-1 xenograft model was established in nude mice to investigate the in vivo effects of the above findings. The results showed that the tumor volume was much smaller in the combination therapy groups than in the single-drug (sorafenib or verteporfin) groups (Fig. 7O, P). Additionally, the results were further evaluated by IHC staining of c-caspase3 and YAP in ex vivo tumor samples (Fig. 7Q). Taken together, sorafenib combined with Verteporfin can reverse the SRGN-induced sorafenib resistance through targeting YAP-activated CSCs.
{kind=link}