HMPV subtyping and subgroup temporal patterns. In total, 232 HMPV G gene sequences were obtained of which 44% (102/232) belonged to subgroup A2 and further clustered into sub-lineages A2a 18% (18/102), A2b 34% (35/102) and A2c 49% (48/102) (Additional file 2). There were no subgroup A1 viruses. Among the sequenced HPMV strains, 56% (130/232) belonged to HMPV group B, of which 82% (107/130) and 18% (23/130) were subgroup B1 and B2, respectively. Multiple subgroups co-circulated in each country (Fig. 1, panel a). Notably, A2a viruses were only identified in South Africa and Zambia. HMPV subgroup temporal patterns in Mali mirrored those in The Gambia (Fig. 1, panel a).
HMPV Intra-country genetic diversity. Only subgroup B1 viruses were detected in high frequencies in all the five countries and were analysed for intra-country diversity (Table 2). ML trees were reconstructed independently for each country. At least two well supported (bootstrap value > 95%) phylogenetic clades were observed in each country (Additional file 3). Sequences from different within-country sampling locations were mixed within the phylogenetic clusters suggesting rapid spread of HMPV variants within each country. Sequences from cases and controls were mixed within the clades (Additional file 3).
HMPV spatial origins and dispersal patterns in Africa. B1 sequences clustered into three major phylogenetic clades, numbered b1C1 to b1C3 (Fig. 2, panel b). Clade b1C2 contained sequences predominantly from South Africa. Sequences from the same geographical region, i.e. West Africa (Mali and Gambia), East Africa (Kenya) and Southern Africa (South Africa and Zambia) closely clustered together (Fig. 2, panel b). On the global MCC tree the three clades (b1C1, b1C2 and b1C3) were placed into two major clades alongside global sequences (Fig. 2, panel a). Clades b1C1 and b1C2 fell into the same clade interspersed with global sequences, and appeared discretely from clade b1C3, suggesting that at least two distinct B1 variants were in circulation (Fig. 2, panel a). The two variants reflect the genetic clusters that were observed on country-specific ML phylogenies above (Additional file 3). Clade b1C1 and b1C2 clustered closely with sequences from Spain and Canada and Malaysia. Clade b1C3 clustered closely with sequences from Malaysia.
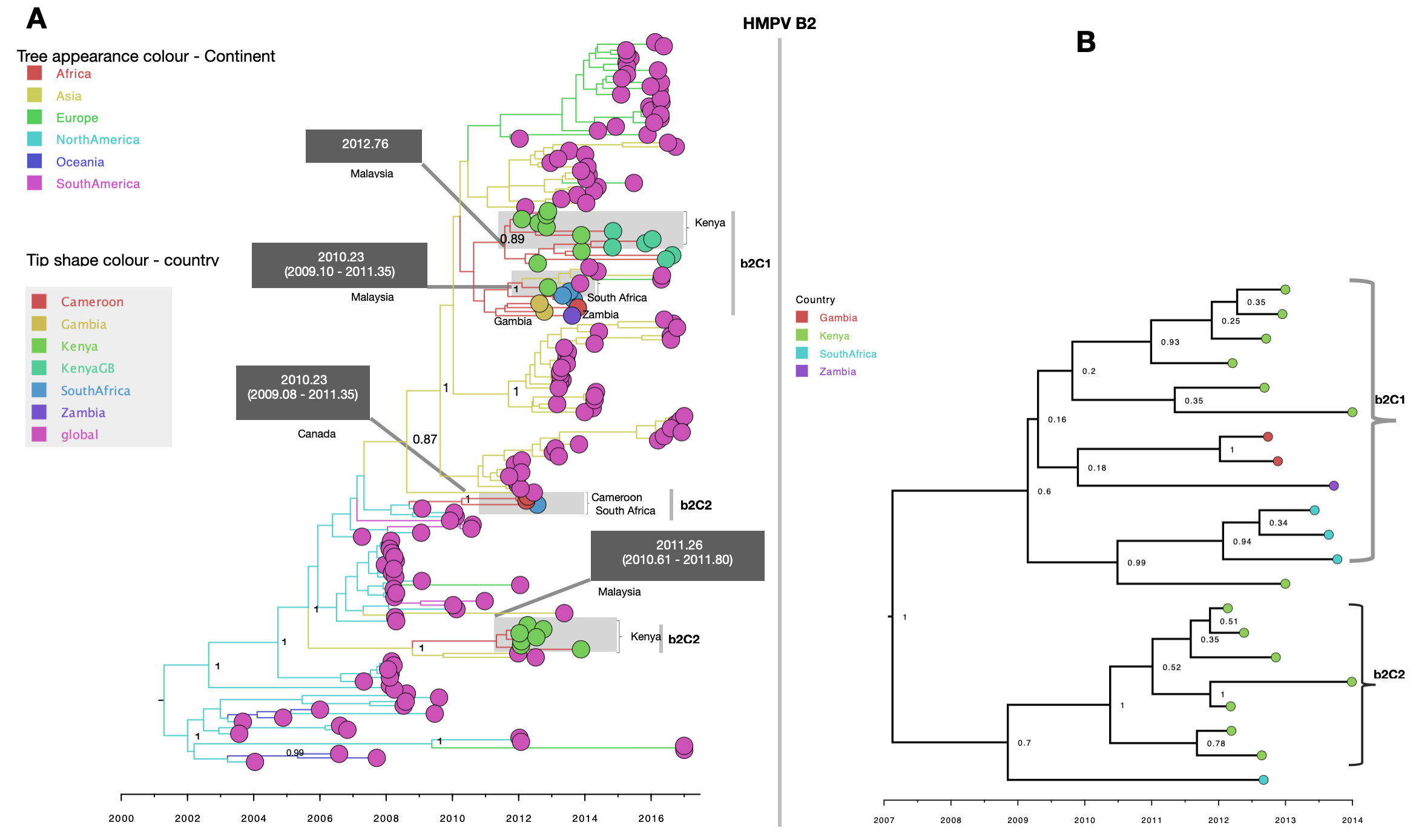
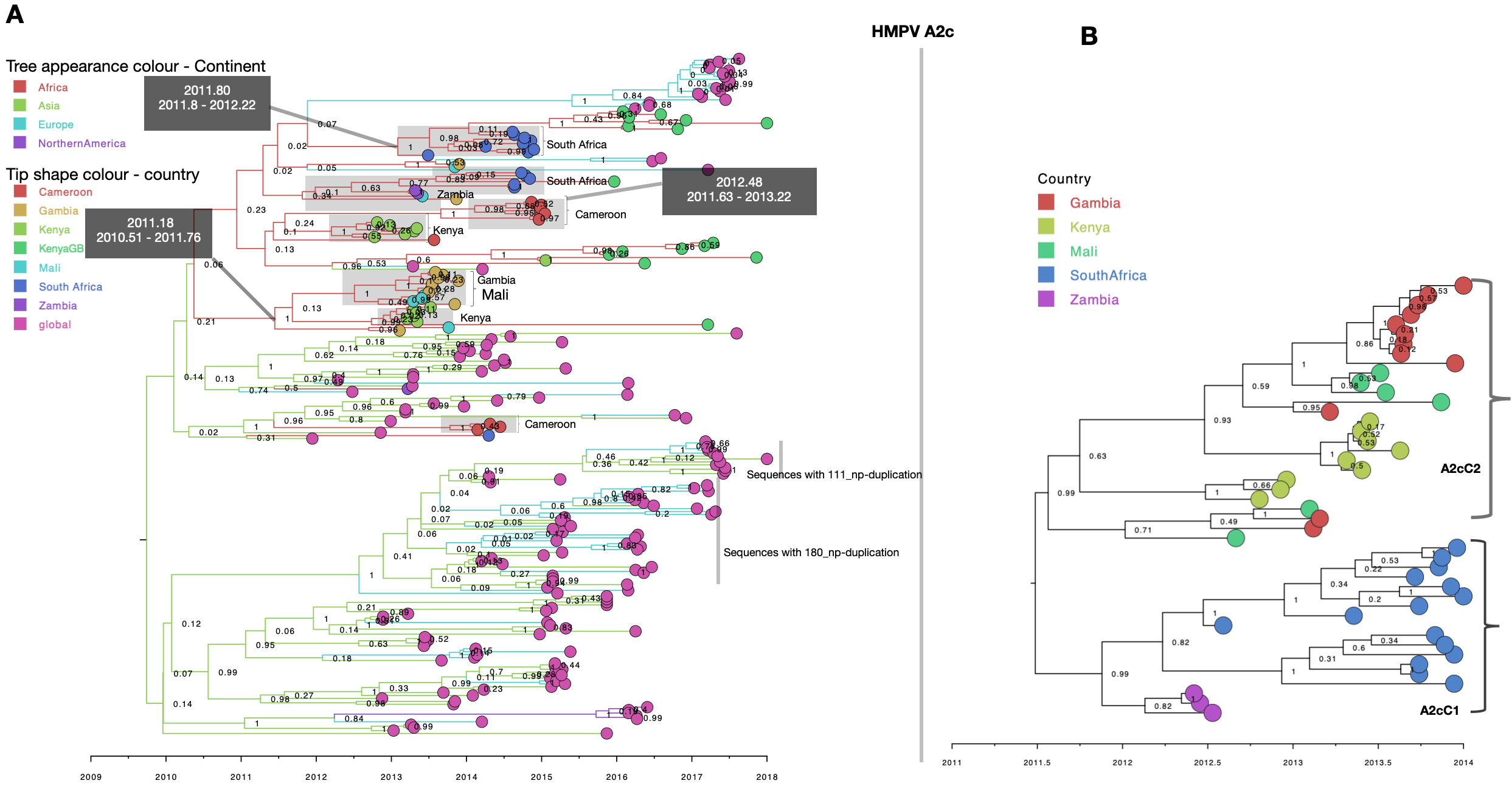
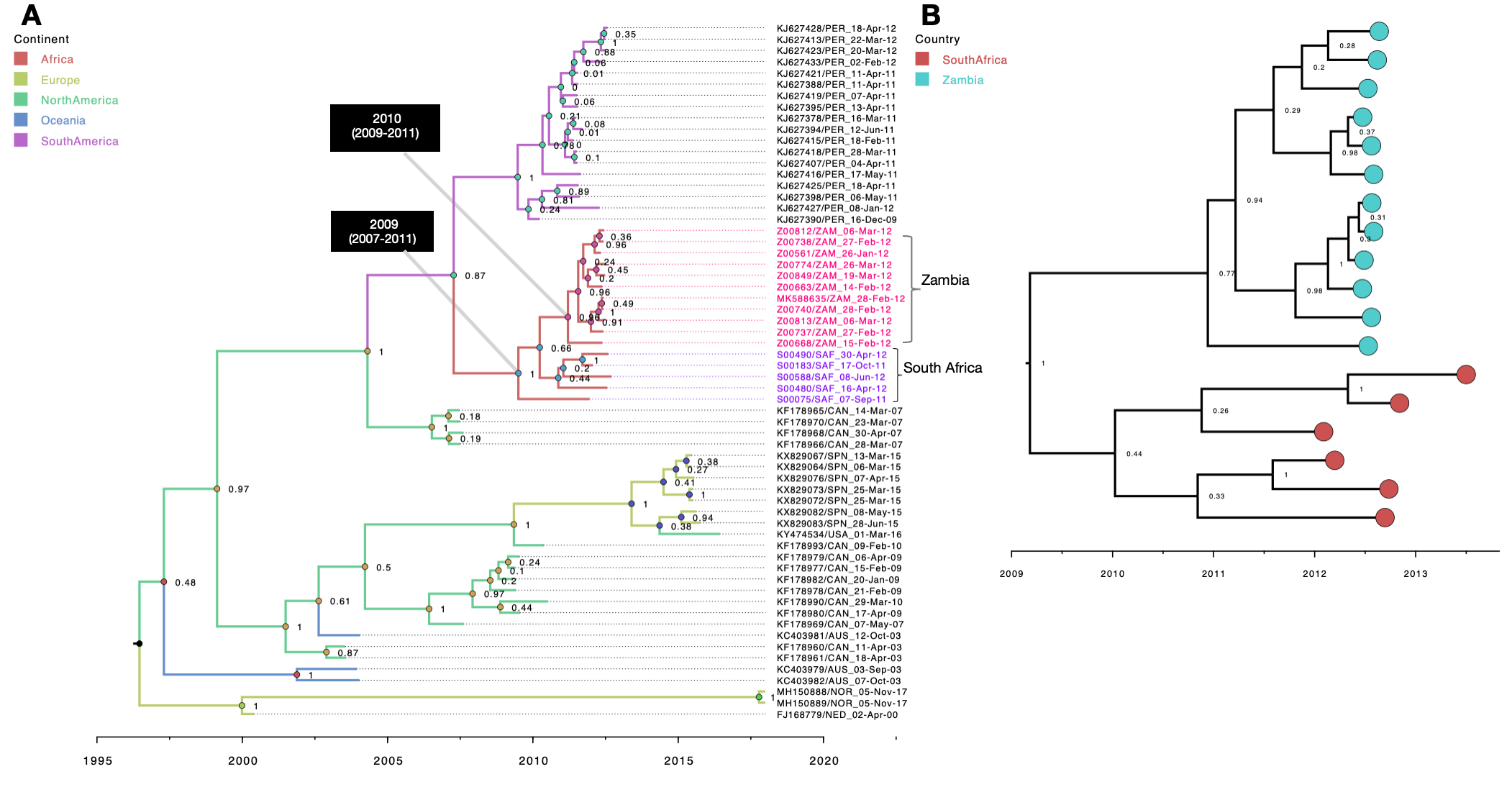
Consistent with B1 MCC phylogenies of A2b, B2 and A2c African sequences showed at least two circulating variants for each subgroup (Fig. 3, Additional files 4 and 5, respectively). Sequences from South Africa and Zambia clustered together. Similarly, sequences from Gambia and Mali clustered more closely among themselves, indicating an epidemiological linkage between neighbouring countries and separate introductions of HMPV variants in Africa. For A2b, sequences clustered into three well supported (posterior probability > 95%) major clades, numbered A2bC1 to A2bC3 (Fig. 3, panel b). On the global phylogeny, the three clades clustered separately interspersed with global sequences (Fig. 3, panel a). Clade A2bC2 and A2bC3 were exclusively made of Kenyan sequences and clustered closely with sequences from Canada (Fig. 3, panel a). Clade A2bC1 contained sequences from South Africa and Zambia and clustered closely with sequences from Peru (Fig. 3, panel a). African B2 viruses clustered at least into two major clades (B2C1 and B2C1) and were placed separately in context of other global B2 sequences suggesting multiple introductions into Africa (Additional file 4). The two clades (B2C1 and B2C1) clustered closely with sequences from Malaysia (Additional file 4). In the A2c time resolved phylogeny, African sequences clustered into two major well supported clades (numbered A2cC1 to A2cC2), see Additional file 5. Clade A2cC1 contained only sequences Zambia and South Africa. On the global A2c MCC tree, the two clades were placed in the same clade and were interspersed with sequences from Spain and Malaysia (Additional file 5). For A2a viruses, African sequences were placed into a single monophyletic clade indicating a single introduction (Additional file 6). Notably, A2a sequences were only detected in Zambia and South Africa and clustered closely with sequences from Peru. On the B1 (Fig. 2) and A2c (Additional file 5) phylogenies, although sequences from Africa were interspersed with global sequences, they mostly clustered together. Of note, 81% (178/228) of B1 and 71% of A2c (165/232) sequences were from Africa and Asia, making it difficult to assess viral introductions from unsampled locations. It is also possible that B1 and A2c viruses could have been prevalent to Africa and Asia.
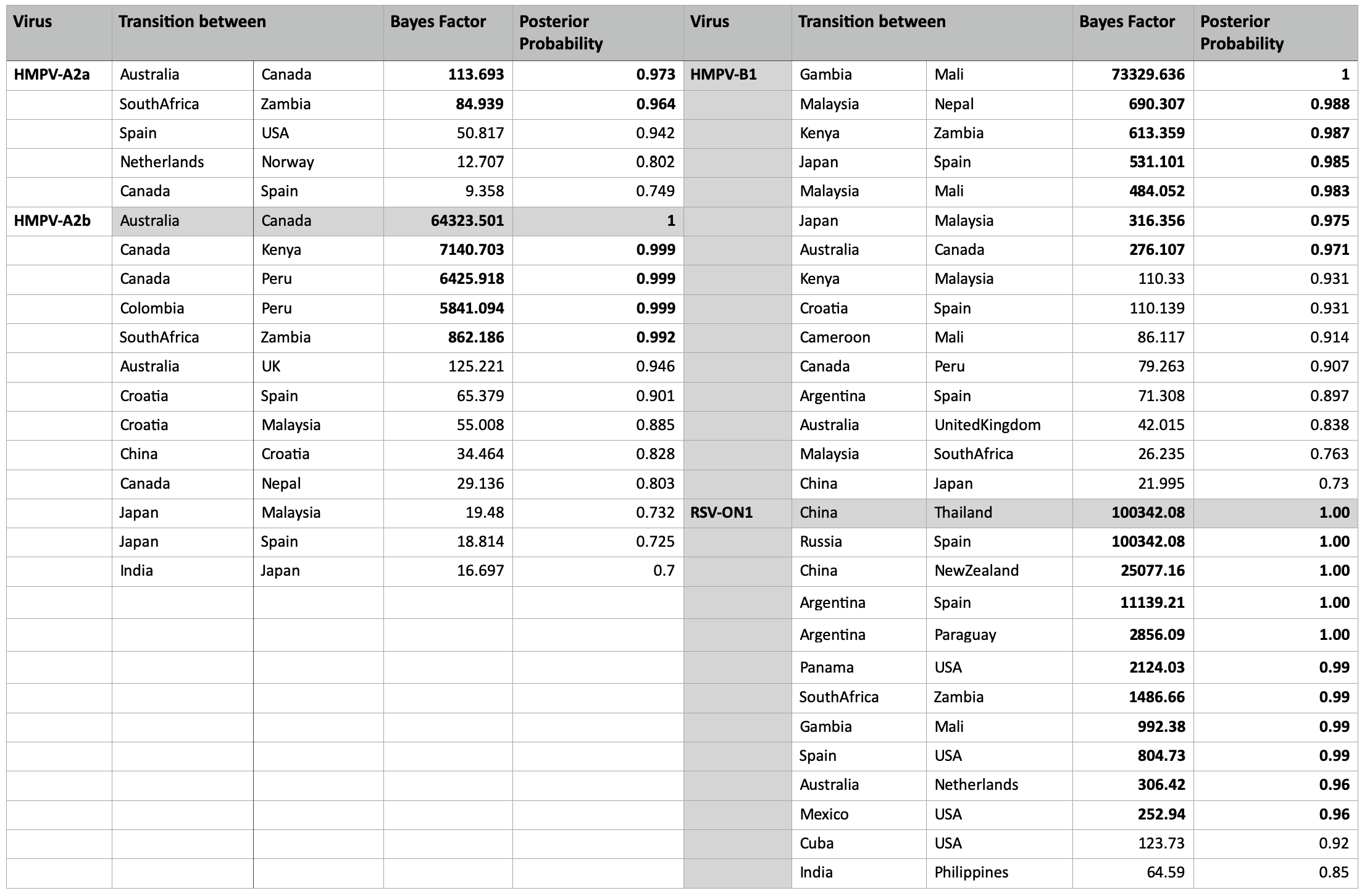
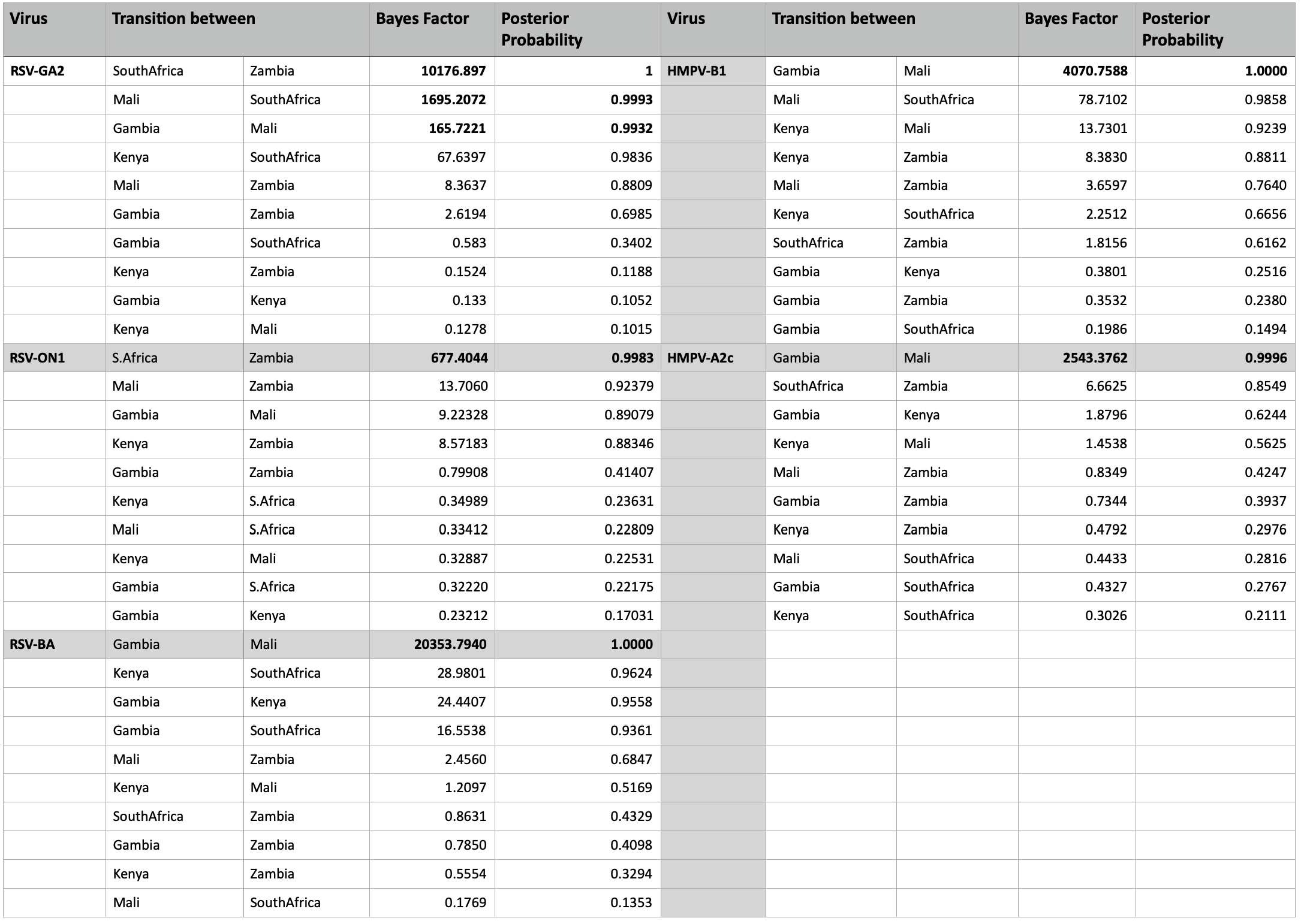
Our phylogeographic analysis indicated global movement of HMPV variants. Very strong (BF > 1000, posterior probability > 95%) and strongly supported (BF > 10, posterior probability > 95%) migration pathways were indicated between different regions globally (Additional file 7). Only migration pathways with BF > 10 and posterior probability > 70% are shown (Additional file 7). Strong connections between close African regions were indicated.
RSV subtyping and subgroup temporal patterns. Based on the G gene phylogeny (Additional file 2), there were 509/842 (60%) RSVA and 333/842 (40%) RSVB sequences. All RSV B sequences belonged to the genotype BA. Among RSV A, 32% (163/509) were genotype ON1, and 68% (346/509) were genotype GA2. Similar to HMPV, multiple RSV genetic groups co-circulated within epidemics (Fig. 1, panel b). Similar genotype dominance patterns were observed between Mali and Gambia, South Africa and Zambia, and were all different from Kenya (Fig. 1, panel b).
RSV intra country diversity. To assess within-country genetic diversity, RSV BA genotype sequences were selected (Table 2). ML trees were reconstructed for each of the five African countries. From the country-specific phylogenies, sequences from the different within-country sampling locations were mixed within the phylogenetic clusters suggesting rapid spread movement of RSV variants within each country (Additional file 8). Similarly, the RSV G gene sequences did not cluster by case or control status of the sampled individuals.
RSV spatial patterns and Origins in Africa. RSV phylogeographic analysis revealed markedly similar spatial patterns to those of HMPV. On the continental scale (Africa), geographical clustering was evident, and multiple variants of each RSV genotype were detected (Figs. 4 and 5). The inferred continental migration pathways indicated very strongly supported links between neighbouring countries (BF > 1000, posterior probability > 95%) i.e., between The Gambia and Mali, and between South Africa and Zambia (Additional file 9). We further explored the RSV spatial patterns globally to elucidate on the viral introductions into Africa. Only RSV genotype ON1 were analysed. African sequences fell into two major clades (numbered C1 to C2, Fig. 6) interspersed with global sequences suggesting at least two distinct variants of RSV ON1 circulated in each of the five African countries. Although the clades C1 and C2 were interspersed with global sequences, high sequence similarity (99%) was observed among them indicating widespread movement of similar variants globally. Of the two African clades (Fig. 6), clade C1 clustered closely with sequences from USA and clade C2 clustered closely to sequences from Spain.
The ON1 global phylogeographic reconstructions indicated viral movements globally although significant links (BF > 100, posterior probability > 95%) were inferred between only a number of pair of regions (Additional file 7). Only locations with migration links of BF > 100 and posterior probability > 70% are reported. Strong connections were still evident between Gambia and Mali, South Africa and Zambia suggesting stronger epidemiological links between close African regions.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}