Recycling organic waste and converting them into renewable energy is a promising route for cleaning environment and effective industrial reactions. This study targeted to isolate a pure anaerobic culture with potential to hydrolyze different biomass and bio-H2 production. For this, a sample of full-scale anaerobic digester, fed with a mixture of biomass was inoculated on Reinforced Clostridial Medium (RCM) in strict anaerobic conditions. An anaerobic Clostridium Butyricum CBT-1 strain was isolated, identified from morphological and 16S rRNA sequence. The strain expressed amylase, cellulase and peroxidases activities. CBT-1 efficiently showed fast growth rate of 3.10 OD/600nm after 72 hours incubation time on Azure B and crystal violet dyes. The strain exhibited 82.4% and 78.5% decolorization of Azure B and crystal violet dyes respectively. In batch fermentation experiment, the CBT-1 produced highest of 3.06, 2.67 and 2.46 mol/mole H2 yield from glucose, starch and cellulose respectively. Whereas, the CBT-1 showed low 0.43 mol/mole from untreated rice straw, and comparatively high H2 yield of 1.91 and 2.01 mol/mole rice straw hydrolysate and kitchen waste respectively. The cumulative volumetric yield of H2 was 358.15, 300.8 and 294.5NmL/gSub from glucose, starch and cellulose respectively. Similarly, the cumulative H2 volume was 76.7, 184.4, 237.2 NmL/gVS from untreated rice straw, rice straw hydrolysate and kitchen food waste. This study emphasizes the prospects to find similar robust anaerobic culture for hydrolyzing complex biomass. Such strains could be used as standard co-inoculum for bio-H2 and biocatalyst for commercial scale applications.
Research Article
Whole Cell of Pure Clostridium Butyricum CBT-1 from Anaerobic Bioreactor Effectively Hydrolyze Agro-Food Waste into Biohydrogen
https://doi.org/10.21203/rs.3.rs-1403093/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 16 Aug, 2022
Read the published version in Environmental Science and Pollution Research →
You are reading this latest preprint version
anaerobic digester
fermentation
Clostridium
biohydrogen
waste biomass
Energy is the basic human need for daily activities. Currently, the world relying on energy of petroleum based fuels generated from fossil sources. The problems with petroleum fuels are releasing of carbon dioxide and increasing of global warming. Further, the fossil sources are depleting quite rapidly and the cost of petroleum based fuels are increasing globally (Roy and Das 2016). To control the price hike and environmental pollution, alternative energy sources have been put on the target (Shah 2017). Renewable energy from cheap carbon sources, robust product specific strains and new bioprocess technological are the possible options (Shah et al. 2018a, Shehbaz 2018). The agricultural waste biomass (grasses, straw) and kitchen waste could be utilize (Wong et al. 2016). The kitchen waste contain mostly, fruits and vegetables, which are easily digestible for the microorganisms (Rodríguez-Valderrama et al. 2020). However, grasses and waste straw needs some pretreatment either chemical, physical or biological to for easy fermentation process (Shah 2018). The reason of pretreatment is to remove the lignin, from waste straw, which acts as a barrier in fermentation process. Lignin removal speed up fermentation and increase the yield from the substrate per gram (Ali et al. 2020). Ideally, this treatment should be nominal and mild to lower the cost and effective for the depolymerization for complex biomass (Shah et al. 2018a). Thus dilute chemicals treatment (H2SO4 acid, NaOH alkali) process can be choose as an inexpensive pretreatment approach (Shah and Tabassum 2018, Shah et al. 2019).
Nature is full of wonderful microorganisms (Shah et al. 2018b, Shah 2019), it only needs to be explore for the screening of target specific bioproduct (Shah et al. 2016b, Aimen et al. 2020). Microorganisms are sources for production of various value added metabolites. In fact, a microbial cell is functioning as a factory, which synthesized different kinds of useful secondary materials. Bacterial species are predominant among other microorganisms with enormous potential of biomolecules production. Previously, we reported pure Bacillus sp. strains capable of conversion of Municipal Organic Food Waste into biohydrogen.
This study further emphasized to screen out more anaerobic fermentative bacteria, which can convert multiple waste biomass into fuels. Anaerobic digestion and anaerobic granular sludge are reported with a wide range of archaea, bacteria and fungi producing hydrogen and methane gas. In the list of these anaerobic microorganisms, Bacillus sp. strains and Clostridium sp. strains are the significant culture inside anaerobic digestion (Collet et al. 2004, Winter 2005). Other culture like enterobacters, Aeromonas, Pseudomonas, Streptomyces and several more isolates are reported (Oh et al. 2003). However, these cultures are either low for H2 potential or non H2 producing. Additionally, these microbes either unable to grow even on simple carbon beside complex biomasses. That does not determine them ideal for H2 production and high scale processing. Any strain capable of converting multiple substrates like starch, cellulose, straw and food waste simultaneously, could be the best culture for commercial applications (Kothari et al. 2010, Hay et al. 2013).
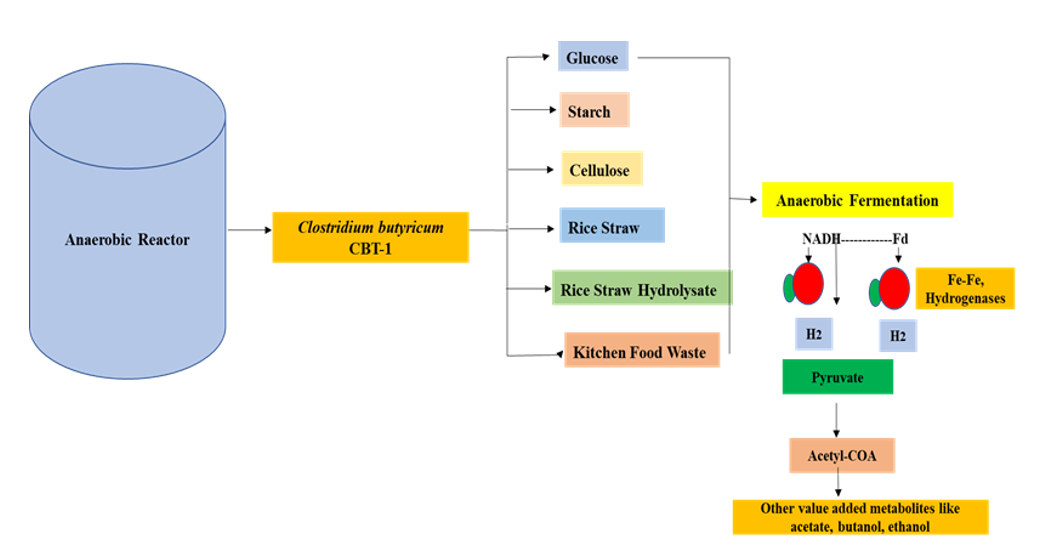
Clostridium butyricum is gram positive, spore forming and one of the highest anaerobic bacterium existing in anaerobic digestion processes. Clostridium butyricum is reported with capabilities to ferment various polysaccharides (Cai et al. 2011, Cai et al. 2013). It can produce a variety of products like acetic acids, butyric acids, solvents like glycerol, butanol and gases (H2). However, many Clostridium strains have been isolated, but only few of them are found effective in fermentation of complex polysaccharides carbohydrates (Cai et al. 2011, Cai et al. 2013). This study focused on bio-H2 production from a pure anaerobic strain capable of hydrolyzing rice straw hydrolysate and kitchen food waste. To get this objective, a granular sludge was collected from an anaerobic reactor actively operational on straw and food waste biomasses. The sample of this anaerobic reactor was screened to isolate the most active and vital Clostridium strains that have the capacity to hydrolyze various biomass including rice straw hydrolysate and kitchen waste. Successfully, Clostridium butyricum CBT-1 strain is purified and demonstrated capabilities of waste biomass hydrolysis. This is our first effort towards purification of Clostridium butyricum CBT-1 for bio-H2 production. It can be stretched to explore more industrial biochemicals of Clostridium butyricum strains. The study emphasize for improvement in batch fermentation process and development of standardized co-inoculum of pure anaerobic Clostridium strains capable of bio-H2 gas production. The experiments were designed as showed in graphical figure below.
2.1 Material Preparation
Most of the chemicals tested in this study were of Sigma Aldrich (Germany). An overnight dried rice straw was grounded by grinding machine to a fine powder of < 0.5 cm size. The rice straw was measured as 40.0% C, 0.95% N, and 4.1% H. The VS and TS composition was 85.3% and 81.5% of the rice straw respectively, representing a high organic matter. The kitchen organic food waste was collected from the University Cafeteria. The unwanted materials like plastic bags, papers, glass and mud were removed. A homogeneous mixture is prepared and the kitchen organic waste samples is stored at 4oC. Rice straw hydrolysate (RSH) was prepared with 1% NaOH at 121ᵒC for 15 min autoclaving. After alkali treatment, the rice straw was subjected to another pretreatment step, this time with 1% H2SO4 at 121ᵒC for 15 min autoclaving. The RSH was filtered, washed with water and dried at room temperature (Minu et al. 2012). The untreated rice straw and rice straw hydrolysate samples were bound with gold palladium on black carbon tape. These samples of untreated rice straw and rice straw hydrolysate were photographed by scanning electron microscope (SEM). SEM images were taken with magnification resolution of 1000 um to see the disrupted fibrous structure of untreated rice straw and rice straw hydrolysate after alkali and acid treatment (Qu et al. 2017).
2.2 Isolation Process, Reinforced Clostridial Medium (RCM)
Anaerobic granular sludge sample was collected in a bucket from an anaerobic reactor fed with organic municipal waste and straw residue after proper mixing to get a homogenized sample. From this bucket, 100 mL samples in triplicates were taken and sealed tightly in 250 mL anaerobic bottles. The 250 mL bottles were kept at 80°C for 3 hr. A 100 mL of Reinforced Clostridial Medium (RCM) compose of meat extract 0.1g/100 mL, starch 0.1g/100 mL, yeast extract 0.3g/100 mL, glucose 0.5g/100 mL, peptones 0.1g/100 mL, sodium acetate 0.3g/100 mL, sodium chloride 0.5g/100 mL, L-cysteinium chloride 0.3g/100 mL, and agar 0.5g/100 mL was sterilized in autoclaved at 121°C for 15 minutes. The RCM bottles were inoculated with 20 mL volume from each granular sludge sample after heat treatment. The RCM bottles were flushed with nitrogen gas for 4 minutes and incubated in the thermal anaerobic chamber at 37°C temperature. After 72hr of growth, fresh sterile RCM broth of 100 mL volume was inoculated with 5 mL from each pregrown RCM bottles. The RCM bottles were incubated again in the thermal anaerobic chamber at 37°C temperature. This time, after 72 hr, the grown culture in RCM bottles was serially diluted in normal saline and 100 uL of culture inoculum from each dilution was spreaded in to sterile RCM agar plates. RCM plates were incubated in strict anaerobic conditions (placed inside an anaerobic chamber equipped with 10% H2, 5% CO2 and 85% N2 gas mixture) at temperature of 37°C. After incubation time for 48 hr culture growth was checked. From the growth culture, a pure single colony was streaked in to fresh nutrient agar plate. RCM media was used for long-term culture growth and spore generation. The pure culture cells was stored at refrigerator temperature (4°C) in sterile semisolid RCM broth tubes and glycerol tubes separately.
2.3 Colony Morphology and Enzyme Qualitative Assay
Spores were picked from the RCM agar plate and activated at 60°C for 15 minutes. Slide smear was prepared. Gram staining was performed to check the morphology of the pure culture under microscope. Also on fresh RCM and nutrient agar plates media, the macroscopic morphology like color, shape, and texture of the colony was observed. Further identification was done by performing biochemical tests using procedure of Bergey’s Manual of Systematic Bacteriology. The pure culture was subjected to qualitative enzymatic study for cellulase and amylase using standard plate method. Pure culture was grown in RCM broth at 37°C, 120 rpm for 24 hr. A 100 µL of cells volume from RCM broth was diluted in 0.9% NaCL. A 3 µL sample of diluted cell suspension was freckled onto media plates in triplicates. For cellulase 5 g/L carboxymethylcellulose (CMC), and for amylase 20 g/L starch was added respectively. The agar plates of CMC and starch were incubated at 37°C for 72 h. The enzyme activity was checked by flooding gram iodine solution for amylase test and 0.1% Congo red solution for cellulase test. The plates were left on room temperature for 30 minutes followed by washing with 1 M NaCl. The zone of hydrolysis on both starch and CMC plates were recorded (Shah et al. 2016b).
2.4 Growth and Degradation Potential of Dyes
CBT-1 pure culture was grown in RCM broth at 37°C, 120 rpm for 24 hr. A 100 µL of freshly grown CBT-1 cells volume from RCM broth was added into sterilized 50 mL bottles of mineral salt media (MSM). The MSM was supplemented with 0.5g/L Azure B and 0.5g/L crystal violet dye in separate bottles as defined previously (Picart et al. 2016, Ravi et al. 2017). Uninoculated media bottles of Azure B dye and Crystal violet dye were used as control media samples. The bottles were kept in shaking incubator conditions of 37ᵒC and 150 rpm for seven days. Daily 2 mL sample was taken to check absorbance at 620nm under UV-visible spectrophotometer for growth observation. Similarly, 5 mL sample before start of experiment and 5 mL samples after completion of seven days experiment were taken. The samples were centrifuged at 10000 rmp for 10 minutes for each sample bottle. The absorbance of supernatant was measured at 651nm for Azure B dye and 592nm for crystal violet dye decolorization, respectively (Picart et al. 2016). The amount of decolorization for both dyes was calculated using the following equation as:

Where Xi = initial absorbance at first day
Xf = final absorbance at last day
2.5 Polymerase Chain Reaction (PCR) of 16S rRNA Gene
A pure colony was picked from RCM agar plate and was grown in sterile Luria Broth (LB Oxiod pH 0.6) at 37°C, 120 rpm for 24 hr. A 2 mL of cells volume from LB broth was centrifuged at 10000rpm for 15 minutes and the supernatant was discarded from the cells pellet. The cells pellet was treated with 300 µL TE Buffer (Tris HCl 10 mM, EDTA 1mM, 1 M NaCl, pH 8.0). A 100 µL of 10% SDS was added to the tubes. The cells pellet was heated at 80°C for 30 minutes. Proteinase K buffer in 1M Tris HCl of 200 µL was added and kept in water bath at 50°C for 60 minutes. A 50µL of 20g RNase was added at room temperature for 60 minutes. Then 200 µL of 6M-NaCl solution and chilled absolute ethanol was added and centrifuged at 10,000rpm for 10 minutes. The supernatant was transferred to other eppendorfs and chilled 1000 µL phenol-chloroform-iso amylalcohol was added again centrifuged at 10000rpm for 10 minutes. The upper most supernatant was wash with 70% chilled ethanol. The cells pellet is dissolved in 100 µL TE buffer. A 1% agarose gel was prepared and the DNA was loaded in 0.5x solution of TBE. The agarose was heated in microwave oven for 1 minute and cooled up to 45°C. A 0.3µg/mL ethidium bromide (Roche, Germany) was added for staining. Electrophoresis was carried out for 1 hour at 80 V. The DNA bands were visualized in UV Transilluminator (UVItec, EEC) and digital photograph was taken. The extracted DNA was then kept at -20 ºC for PCR reaction. The 16S rRNA region was amplified with forward primer FD1 (5/CCGAATTCGTCGACAACAGAGTTTGATCCTGGCTCAG3/) and reverse primer RD1 primer (5/CCCGGGATCCAAGCTTAAGGAGGTGATCCAGCC3/). Taq DNA polymerase (Fermentas, USA) of 1.5 µL, 1.5µL of 50ng genomic DNA, 15µL 10x PCR Super mix, 1.5µL from (25ng/µL) forward primer and reverse primer were added. PCR buffer water of 11.5µL were added to PCR tubes and gently vortexed for 5 seconds. PCR conditions were set as such : initial denaturation of one time at 94°C for 2 min, 94°C for 1 minute in each cycle, extension at 72°C for 1 minute and annealing at 55°C for 1 minute is processed for 30 PCR cycles. The PCR product was cleaned with QIAquick PCR Purification Kit (Qiagen, MD, USA). The amplified PCR sample was sequenced. The raw sequence of PCR was filtered through the sequence analysis package (DNA-Star). Fasta sequence of 16S rRNA was searched through NCBI Basic Local Alignment Search Tool (BLAST) for genetically similar species strains. Phylogenetic tree was constructed using MEGA7 software(Shah et al. 2019).
2.6 Batch Fermentation for Biohydrogen Potential (BFBP) from Glucose, Starch and Cellulose
Glucose, starch and cellulose 15 g/L was added into sodium phosphate buffer (SPB) of pH 6.5 supplemented with K2HPO4 (2.5 g/L), (2.5 g/L) NaHCO3 solution, 2 mL of vitamin solution in 250 mL Pyrex bottles. The volume of media was kept to 100 mL equally in all Pyrex bottle. The pH was balanced at 6.5 for each Pyrex bottle. Then media was autoclaved for 15 min at 121° C. Pure colony of CBT-1 was anaerobically grown in sterilized LB broth at 37°C overnight. The media Pyrex bottles containing 15 g/L glucose, starch and cellulose separately were inoculated. A starting value of CBT-1 with 0.3nm optical density (600 nm) was equally added into all triplicates bottles. Control (uninoculated) media Pyrex bottles in triplicates containing 15 g/L glucose, starch and cellulose without culture inoculation were run in parallel at the same conditions. All media Pyrex bottles were closed using a silicon plug. Anaerobic conditions were adjusted by N2 gas flushing for 4 minutes in all experimental bottles. The BFBP experimental Pyrex bottles were incubated at 37°C in a thermostatic chamber at static condition for 14 days. The daily volume of biohydrogen produced was measured using water displacement process. A 25% acidified (pH < 3) NaCL solution of 0.5 Liter flask was prepared to record the volume of daily gas released from the headspace of each Pyrex bottle. The amount of water move in graduated cylinder is correspondingly equal to the amount of gas released from the headspace of each Pyrex bottle. The hydrogen, carbon dioxide composition were measured by Gas chromatography (micro-GC Varian 490GC).
2.7 BFBP from Organic Food Waste and Rice Straw Hydrolysate
After simple substrates (cellulose, glucose and starch) BFBP confirmation experiments, the culture of CBT-1 was assessed for capability of bioH2 production from complex substrates. The CBT-1 was freshly grown in 100 mL Pyrex bottle of LB medium at 37°C, for 48hr in a thermostatic incubator. Sodium phosphate buffer (SPB) of pH 6.0 supplemented with K2HPO4 (2.5 g/L), (2.5 g/L) NaHCO3 solution, 2 mL of vitamin solution, and 15 g/L rice straw hydrlysate, 15 g/L untreated rice straw and 15 g/L VS of kitchen food waste were prepared for media. A total of 100 mL media volume was kept in each 250 mL Pyrex bottle. Control Pyrex bottles added with untreated rice straw, rice straw hydrolysate and kitchen food waste were run in parallel. All Pyrex bottles were autoclaved for 15 min at 121°C. A 0.3nm optical density of CBT-1 was inoculated in the rice straw hydrlysate, untreated rice straw and kitchen food waste Pyrex bottles. The control Pyrex bottles were left uninoculated. Each Pyrex bottle was flushed for 5 min with N2 gas. All the samples were managed in triplicate. The batch fermentation was run for 14 days at static condition by incubating in 37°C thermostatic chamber. The daily volume of biohydrogen produced was measured using water displacement process as described above in 2.6 section.
2.8 Kinetic Calculations
The kinetic for hydrogen production rate and yield was measured as did earlier in our study (Shah et al. 2016a).The volume of gas produced in the control Pyrex bottle was subtracted from the gas volume in inoculated Pyrex bottle to calculate actual gas yield. The Microsoft Excel program was used to calculate volume of hydrogen (H2), volume of headspace, concentration of H2 at time t and t-1. Whereas, concentration (X) of total H2 volume at time t and the specific H2 concentration at time t and t-1. The daily H2 volume raw data was normalized at standard temperature and pressure (STP). The cumulative H2 volume of each substrate in mL/gVS and mol/mol yield was mathematically calculated using the equation Eq-2 from the daily (H2) volume and concentrations.
\(H2Vol.t=XH2,GH2V,t+HSV.(XH2,t - XH2,t - 1)\) (Eq-2)
Total volume of H2 in mL = H2 Vol, t,
Concentration of H2 = XH2
Gas volume of H2 each time = G H2V, t
Headspace volume = HSV
The H2 yield was calculated as shown in the equation Eq-3.
\(H2{\text{ }}(Y)=\frac{{Cumulative{\text{ }}H2}}{{Weight{\text{ }}of{\text{ }}substrate}}\) (Eq-3)
Where H2 yield (Y) = measured as mL/gVS as substrate load was based on VS to each Pyrex bottle in BHFP.
The H2 yield (Y) mol H2/g glucose was measured by equation Eq. 4.
\(H2(y)=\frac{{Cumulative{\text{ }}H2}}{{\frac{{22.4}}{{\frac{{Weight{\text{ }}of{\text{ }}glu\cos e}}{{180}}}}}}\) (Eq-4)
where ,
1 mole of ideal gas at standard temperature and pressure (STP) = 22.4 L/mol volume
1 mole of glucose = 180 g/mol
The estimated kinetic of total hydrogen potential was measure by modified Gompertz Model (Eq. 5) by Statistical software (IBM SPSS Statistic 23) through nonlinear regression model for each sample separately and the values of P, R and L were calculated.
\(Y\left( t \right)=P \times {\text{ }}exp{\text{ }}\left\{ { - {\text{ }}exp{\text{ }}\left[ \begin{gathered} \underline {{{R_*}^{e}}} \hfill \\ p\left( {\Delta - t} \right) \hfill \\ \end{gathered} \right]{\text{ }}\left( {L - t} \right)+1} \right\}\) (Eq. 5)
Where Y (t) is total yield of hydrogen (mL) in total time of incubation (t), P is the hydrogen production (mL), R is highest rate of production (mL/d), and L is the lag phase time in days (d), e is equal to 2.718282.
2.9 Analytical methods
Rice straw and kitchen food waste was analyzed for carbohydrate composition. Total solids (TS), volatile solids (VS), ash, moisture, carbon content, nitrogen, lignin, glucan, xylose, galactose, mannose and arabinose were measured as described in the standard laboratory analytical procedure (LAP) (Sluiter et al. 2013). The TS measurement was calculated by taking 1 gram of rice straw and kitchen food waste, oven dried at 105°C overnight in a crucible. The weight of oven dried rice straw and kitchen food waste was measured again and TS value was calculated using this Eq. 6:
TS (%) = \(\frac{{WSD - WD}}{{DS - WD}}\) (Eq. 6)
Where, WSD = Weight of dried residue + dish, WD = Weight of dish, DS = Dish + substrate.
The VS of rice straw and kitchen food waste was calculated by burning the oven dried rice straw and kitchen food waste samples at 550°C for 30 minutes. The samples of rice straw and kitchen food waste were cooled down in a desiccator at room temperature. The difference in measured weight was found using Eq. 7:
VS (%) = \(\frac{{WDR - WA}}{{DS - WA}}\) (Eq. 7)
Where: WDR = Weight of dried residue + dish, WA = Weight of ash, DS = Dish + substrate.
Rice straw sample was acid treated using the National Renewable Energy Laboratory (NREL)’s analytical method. Carbohydrate monomers (glucan, xylose, galactose, mannose and arabinose) were measured by HPLC (Shimadzu, SPD-MZ0A). Samples before fermentation and post-fermentation experiments of glucose, cellulose, starch, rice straw, rice straw hydrolysate and kitchen food waste were collected for VFA analysis. For carbohydrates detection, a standard solution of (H20 and methanol) were run as carrier at a speed reaction of 0.6 mL/min and 80°C. The filtered samples before and after completion of batch fermentation assay, were treated with phosphoric acid (H3PO4). The samples were run in HPLC (C18 column, mobile phase 1:1) parallel to standard concentrations of ethanol, methanol, n-butyric, propionic, acetic, and valaric acid (Shah et al. 2016a).
3.1 Selection of Targeted Strain
A sample from anaerobic reactor was grown on RCM to enriched only anaerobic culture. Out of four different culture isolates, only one colony was picked that showed qualitative expression for peroxidases, amylase and cellulase potential. The DNA was extracted from pure colony of this isolate named as CBT-1. Primers FD1 and RD1 were used to amplify the 16S rRNA gene by PCR. From PCR sequence and NCBI Blast result, 16S rRNA sequence of the pure CBT-1 culture showed 99.1% homology with Clostridium butyricum strains. The sequence of CBT-1 has been assigned with accession number OM698377 in NCBI Gene submission programme. The similarity pattern of this strain was made through MEGA 7.0 by neighbor joining method as showed in phylogenetic tree (Fig-1). The CBT-1 strain demonstrated its homology with other anaerobic Clostridium strains as well.
3.2 Characterization of the Strain
A total of four different pure microbial colonies were picked from the RCM agar plates based on morphology differences. These colonies were screened for peroxidases, amylases, and cellulases activities by qualitative hydrolysis of Azure B dye, Crystal Violet dye, starch, and cellulose as sole carbon source respectively. Out of four cell colonies, only the CBT-1 isolate showed growth potential and decolorization of Azure B and Crystal violet dyes. CBT-1 maximum growth was observed at 4th day of incubation time. The growth-started decline after 6th day as showed in (Fig. 2). The growth pattern of CBT-1 is similar to previously reported studies of bacterial culture on similar dyes (Chandra et al. 2007, Abd-Elsalam and El-Hanafy 2009).
In Fig. 3, (A,B) images it was found that the CBT-1 strain is a Gram positive, rod shaped, spore forming, and mesophilic anaerobic culture. Colony of the CBT-1 was convex, lobate and off white on LB agar medium at 37°C for 48 hr. In the gram staining (Fig. 3, A) slide smear, its cells appeared as straight rods occurring in single or pairs (Fig. 3). The CBT-1 also showed zone of hydrolysis on both CMC and starch plate, indicated positive for its enzyme activities (amylases and cellulases) as shown in the Fig-3. In blue medium (C) in Fig. 3, a zone around colonies showed starch hydrolyzing activity. Similarly, in reddish medium (D) in Fig. 3, a zone around colonies showed existence of cellulolytic activity. The ability of the CBT-1 strain to grow on starch and CMC as a carbon source and formation of zone around colonies confirmed that they could metabolize it into product. Also can hydrolyse other biomass containing cellulase and starch composition. The CBT-1 growth ability confirmed its degradation efficiency of both dyes. It was found 82.4% decolorization of Azure B dye and 78.5% of crystal violet dye after seven days of incubation respectively (Fig. 3). Previously, we also observed decolorization of Azure B dye using pure culture of Bacillus sp. strains (Shah 2014). These pure cultures of Bacillus sp. strains were producing peroxidase (ligninases, laccase). The investigation of dyes degradation potential was for the validation of enzymes system and expression of ligninases, laccase, and plate assay of CMC for cellulases, and starch for amylase. This strategy helps in confirmation of lignocellulosic and kitchen food waste digestion for bio H2 production. This strain was selected for further analysis.
3.3 Substrate Composition
Rice straw and kitchen waste was analyzed for its compositional analysis. The rice straw was containing 20.2% lignin, total organic carbon 51.5%, total nitrogen 0.55%, total solid 92.5% and volatile solid 85%. Whereas, the kitchen waste was containing 5.2% lignin, total organic carbon 45.7%, total nitrogen 1.5%, total solid 16.5% and volatile solid 78.6%. The detail composition of substrates shown in the (Table 1). The compositions of rice straw is consisted with earlier described results (Shah et al. 2019).
|
Parameters |
Rice Straw |
Rice Straw Hydrolysate |
Kitchen Food Waste |
|---|---|---|---|
|
Moister |
5.5 |
6.2 |
70.5 |
|
Ash |
9.3 |
5.5 |
12.3 |
|
Glucan |
41.5 |
38.6 |
8.1 |
|
Xylan |
21.2 |
17.5 |
6.4 |
|
Arabinan |
2.1 |
1.0 |
0.3 |
|
Galactan |
1.3 |
0.5 |
NP |
|
Manan |
NP |
NP |
NP |
|
Lignin |
20.2 |
4.5 |
5.2 |
|
Total solid |
92.5 |
83.0 |
16.5 |
|
Volatile solid |
85.0 |
80.4 |
78.6 |
|
TOC |
51.5 |
49.2 |
45.7 |
|
TN |
0.55 |
0.35 |
1.5 |
| (NP = not present, TN = total nitrogen, TOC = total organic carbon) | |||
Further, the rice straw was treated with acid and NaOH. The effect of treatment was checked for the untreated rice straw, NaOH treated rice straw and after both treatment of 1% NaOH and 1%H2SO4 acid. The SEM images make it obvious that the untreated rice straw micrograph was smooth, flat, and very compact structure (Fig-4-A). After the NaOH hydrolysis, the compact structure of rice straw was degraded (Fig-4-B). The impact was even more severe after both NaOH and H2SO4 acid treatment. This could be due the removal of basic barrier element lignin from the surface of rice straw along with hemicellulose component, showed in (Fig-4-C). The SEM images of Fig-4 clearly revealed breaks in the silicon waxy morphology of the rice straw. Therefore, the rice straw hydrolysate was containing less lignin then as rice straw. Similar changes in surface destruction of the lignocellulosic biomass due to pretreatment is also reported (Zeng et al. 2011).
3.4 Daily and Cumulative Hydrogen Yield
The Clostridium species consist of huge number of extracellular enzymatic system (cellulosomes) necessary for hydrolysis of complex biomass. Several Clostridium species are reported for utilization of pure carbohydrates (i.e., sucrose, glucose, and galactose), polysaccharides enriched municipal waste and lignocellulosic biomass comprise of hemicelluloses and celluloses. The efficient utilization of lignocellulosic biomass require a pretreatment step to convert the substrate into sugar units for easy uptake and fast fermentation process (Latifi et al. 2019). This study first confirm the extracellular enzymes (peroxidase, cellulases, and amylases) and then tested for biohydrogen production from pure carbohydrates. Rice straw hydrolysate was included, which could be an encouraging step towards the use of plant biomass for the synthesis of bioH2 and other valuable products.
As per objective of the study, the culture of Clostridium Butyricum CBT-1 was first evaluated for H2 production from glucose, cellulose and starch. It was found that the Clostridium Butyricum CBT-1 released H2 with similar yields from starch and cellulose, whereas slightly different in case of glucose as carbon source. Glucose gives comparatively higher yield than cellulose and starch. The Clostridium Butyricum CBT-1 produced H2 yield of 3.06, 2.67 and 2.46 mol/mole of consumed glucose, cellulose and starch respectively. Likewise, Clostridium Butyricum CBT-1 produced H2 yield of 0.43, 1.91, 2.01 mol-H2/mol of consumed untreated straw, rice straw hydrolysate and KFWS respectively. Similarly, the daily volumetric rate of H2 production from glucose to starch and cellulose was noted. The daily highest H2 volumetric rate was 48 mL H2/d from glucose, 43.3 mL H2/d from starch and 41.5 mL H2/d from cellulose respectively. Similarly, the cumulative volume of H2 potential was 358.15 NmLH2/substrate from glucose, 300.8 NmLH2/substrate from starch and 294.5 NmLH2/substrate from cellulose respectively as showed in (Fig. 5). The hydrogen yield of this study is in line with previous reported H2 yield from similar substrates like glucose, starch, and cellulose (Xing et al. 2009). Recombinant C. thermocellum DSM 1313 is reported for H2 production from cellulose and hemicellulose substrates, which suggested that pure culture, can use plant waste straw for value added products (Xiong et al. 2018). A co-culture of E. aerogenes and C. butyricum showed a 2mol H2/mol glucose production rate from starch waste and corn (Yokoi et al. 1998). Whereas, the highest rate H2 yield of 3-3.8 mol H2/mol hexose, have been observed from Pyrococcus furiosus, Thermotoga spp, and Thermoanaerobacterium spp (Verhaart et al. 2010).
The (Fig. 6) illustrates the daily and cumulative H2 production from untreated rice straw, rice straw hydrolysate and kitchen food waste. The daily highest H2 volume was 11 NmL/gVS from untreated rice straw, 30.2 NmL/gVS from rice straw hydrolysate and 34.1 NmL/gVS from kitchen food waste. The maximum cumulative volume of H2 produced was only 76.7 NmL/gVS from untreated rice straw, 184.4 NmL/gVS from rice straw hydrolysate and 237.2 NmL/gVS from kitchen food waste respectively.
No methane gas was found from the batch fermentation of all the substrates (glucose, starch, cellulose, rice straw and kitchen food waste fermentation experiments. The concentration of H2 detected in GC was in the range 25–40% for the tested substrates. The outcomes of the present study are comparable to previously reported cumulative H2 yield from similar biomass and carbon carbohydrate saccharides (Xing et al. 2009, Shah et al. 2016b).
3.5 Advantages and Comparison of Pure culture for Hydrogen Production
In this study, the CBT-1 strain total hydrogen yield was compared as showed in (Fig. 7). It was found that he highest H2 yield was exhibited 393.18 NmL/gVS from glucose. Whereas, the cellulose and starch comparatively produced similar H2 yield was exhibited 314.18 and 325.62 NmL/gVS respectively. The untreated rice straw, rice straw hydrolysate and Kitchen food waste showed 78.4, 197.4 and 263.3 NmL/gVS H2 yield respectively as showed in Table 2. In (Fig. 8), a comparison of the experimental cumulative H2 volume and theoretical cumulative volume from glucose in 14 days is shown. The experimental H2 volume is relatively similar to theoretically estimated H2 volume of the glucose. Similar H2 volume was observed for other substrates too (data not showed). The overall results of Clostridium Butyricum CBT-1 strain are notable, because only few studies reported biohydrogen potential from a pure culture. However, the capability of H2 potential from lignocellulosic biomass (rice straw and rice straw hydrolysate in this study) is more interesting. The H2 yield from rice straw by Clostridium Butyricum CBT-1 is higher than Ca(OH)2 and acid treated biomass straw as reported previously (Nasirian et al. 2011, Reilly et al. 2014). The chemicals treatment are used to enhanced the hydrogen yield from waste biomass (Reginatto and Antônio 2015). As a 68.1 ml H2/g TVS cumulative H2 yield was obtained from HCl treated agriculture straw (Fan et al. 2006). Similarly, from untreated cornstalk waste a 61.4 mL/g of cumulative H2 yield was observed using Clostridium thermocellum as a single culture. Even, Clostridium thermocellum and Clostridium thermosaccharolyticum co-culture produced 75 mL of H2/g H2 yield (Li and Liu 2012, Li et al. 2012). This is noteworthy, that most of the studies either used mix microorganism of anaerobic sludge as inoculum or expensive chemothermal pretreatments. As, H2 production potential of Clostridium Saccharolyticus from wheat straw hydrolysate treated at 130°C for 30 min has been reported (Ivanova et al. 2009).
In addition, the hydrogen yield of Clostridium Butyricum CBT-1 is higher than previously reported studies. The application and H2 yield of Clostridium Butyricum CBT-1 proved that pure active and biomass degrading culture can avoid the necessities of expensive chemicals pretreatment of waste straw carbon sources. Clostridium Butyricum CBT-1 showed overall 357.15, 293.5, 299.8, 184.4 and 236.2% more hydrogen yield compared to untreated rice straw sample. In our previous study, pure ligninolytic culture Brevibacillus agri AN-3 showed 293.7% increase of hydrogen yield from wheat straw compared to untreated wheat straw sample (Shah et al. 2018a). The bioH2 production is directly proportional to the concentration of VFA consumed during anaerobic digestion process. The level of VFA produced and consumed reflected the performance of substrates fermentation assay and elucidate the H2 metabolic pathways (Luo et al. 2019). In this study, no VFAs concentrations were detected at the completion of glucose, cellulose and starch fermentation assay, signifying the total consumption of VFA to products by the tested Clostridium Butyricum CBT-1. However, in sample of rice straw hydrolysate and kitchen food waste, a considerable level of VFA were detected. The higher concentration was of butyrate, iso-butyrate, acetate, and propionate. The detection of butyrate and acetate VFA production in Clostridium Butyricum CBT-1 proved it ideal candidate for bioH2 production. Similar results of VFA (butyrate and acetate) is reported in the H2 producing microorganisms specifying similar H2 catabolic pathways (Shin et al. 2004, Fang et al. 2006). The leftover VFA and slurry after anaerobic fermentation process is a rich source of beneficial nutrients for plants, crops and vegetables growth. Which can be use as an organic fertilzers to enhance their growth and yield (Hamid et al. 2021). The overall results of this study highlight the importance of isolation of pure culture capable of different biomass hydrolysis and fermentation into value added metabolites. This strategy can be used to collect specific strains for pure bioproducts as well as a biocatalyst for hydrolysis of complex biomass at large scale.
|
P |
L |
Rmax |
R2 |
|
|---|---|---|---|---|
|
Glucose |
393.18 |
1.69 |
40.6 |
0.997 |
|
Cellulose |
314.79 |
1.606 |
37.24 |
0.998 |
|
Starch |
325.62 |
1.602 |
36.32 |
0.998 |
|
KFWS |
263.3 |
2.23 |
29.7 |
0.997 |
|
RS Hydrolysate |
197.4 |
1.85 |
25.5 |
0.998 |
|
Untreated RS |
78.4 |
4.01 |
10.6 |
0.989 |
In this study, Clostridium Butyricum CBT-1 strain was isolated and purified that was capable of expressing amylase, cellulase and peroxidases enzymes. These enzymes activities were determined from a significant (82.4 and 78.5%) amount of decolorization of azure B and crystal violet dyes. In addition, the strain showed considerably high H2 potential from glucose, starch, cellulose, KFWS and rice straw hydrolysate. Especially, the Clostridium Butyricum CBT-1 cumulative H2 yield was significant from rice straw hydrolysate and kitchen food waste. It produced 180–240% more H2 volume from rice straw hydrolysate and kitchen food waste compared to the untreated rice straw. These results indicates a promising line for enhancing H2 yield of pure anaerobic culture from organic waste biomasses. Although, culture conditions optimization may also play an important role to uplift the yield of specific product from similar anaerobic culture.
Authors contribution
Tawaf Ali Shah did investigation, conceptualization, experiments, writing and manuscript preparation, L Zhiyu, and Andong Zhang helped in writing and manuscript preparation Li Zhihe provide supervision and editing, Di Lu, did results and samples analysis; Wang Fan and H. Xuan did samples analysis, equipment and resources management.
Funding
This work was supported by the National Key R&D Program of China (Grant No. 2019YFD1100600).
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
Ethics approval and consent to participate
This study does not involve any humans or animals during experimentation, so it is not applicable in this study.
Consent for publication
This study does not contain data from any individual person.
Competing interests
The authors declare no competing interests.
- Abd-Elsalam HE, El-Hanafy (2009) Lignin biodegradation with ligninolytic bacterial strain and comparison of Bacillus subtilis and Bacillus sp. isolated from Egyptian soil. Am Eurasian J Agric Environ Sci 5:39–44
- Ali ST, Raheem U, Mustafa M, and Rashida (2020) Reprocessing of NaOH black liquor for pre-treatment of agribiomass. Intl J of Agri Sci and vet med 8:1–10
- Cai G, Jin B, Monis P, Saint C (2013) A genetic and metabolic approach to redirection of biochemical pathways of Clostridium butyricum for enhancing hydrogen production. Biotechnol and bioenginer 110:338–342
- Cai G, Jin B, Saint C, Monis P (2011) Genetic manipulation of butyrate formation pathways in Clostridium butyricum. J of Biotechnol 155:269–274
- Chandra R, Raj A, Purohit H, Kapley A (2007) Characterisation and optimisation of three potential aerobic bacterial strains for kraft lignin degradation from pulp paper waste. Chemosphere 67:839–846
- Collet C, Adler N, Schwitzguébel JP, Péringer P (2004) Hydrogen production by Clostridium thermolacticum during continuous fermentation of lactose. Intl J of Hydro Energ 29:1479–1485
- Fan YT, Zhang H, Zhang SF, Hou HW, Ren BZ (2006) Efficient conversion of wheat straw wastes into biohydrogen gas by cow dung compost. Bioresour Technol 97:500–505
- Fang H, Li C, Zhang T (2006) Acidophilic biohydrogen production from rice slurry. Intl J of hydro Energ 31:683–692
- Hamid S, Ahmad I, Akhtar MJ, Iqbal MN, Shakir M, Tahir M, Rasool A, Sattar A, Khalid M, Ditta A (2021) Bacillus subtilis Y16 and biogas slurry enhanced potassium to sodium ratio and physiology of sunflower (Helianthus annuus L.) to mitigate salt stress. Environ Sci and Pollut Res 28:38637–38647
- Hay JX, Wu J, Juan C, Jahim J (2013) Biohydrogen production through photo fermentation or dark fermentation using waste as a substrate overview, economics and future prospects of hydrogen usage. Biofuel Bioprod and Biorefin 7:334–352
- Ivanova G, Rákhely G, Kovács KL (2009) Thermophilic biohydrogen production from energy plants by Caldicellulosiruptor saccharolyticus and comparison with related studies. Intl J of Hydrog Energ 34:3659–3670
- Kothari R, Tyagi V, Pathak A (2010) Waste-to-energy, A way from renewable energy sources to sustainable development. Renewabl and Sustain Energ Rev 14:3164–3170
- Latifi A, Avilan L, Brugna M (2019) Clostridial whole cell and enzyme systems for hydrogen production: current state and perspectives. Appl Microbiol and Biotechnol 103:567–575
- Li Q, Liu CZ (2012) Co-culture of Clostridium thermocellum and Clostridium thermosaccharolyticum for enhancing hydrogen production via thermophilic fermentation of cornstalk waste. Intl J of Hydrog Energ 37:10648–10654
- Li YC, Liu YF, Chu CY, Chang PL, Hsu CW, Lin PJ, Wu SY (20120 Techno-economic evaluation of biohydrogen production from wastewater and agricultural waste.Intl J of Hydrog Energ37:15704–15710
- Luo K, Pang Y, Yang Q, Wang D, Li X, Lei M, Huang Q (2019) A critical review of volatile fatty acids produced from waste activated sludge: enhanced strategies and its applications. Enviro Sci and Pollut Res 26:13984–13998
- Minu K, Jiby KK, Kishore V (2012) Isolation and purification of lignin and silica from the black liquor generated during the production of bioethanol from rice straw. Biom and Bioenerg 39:210–217
- Nasirian N, Almassi M, Minaei S, Widmann R (2011) Development of a method for biohydrogen production from wheat straw by dark fermentation. Intl J of Hydrog Energ 36:411–420
- Oh YK, Park MS, Seol EH, Lee SJ, Park S (2003) Isolation of hydrogen-producing bacteria from granular sludge of an upflow anaerobic sludge blanket reactor. Biotechnol and Bioproc Engineer 8:54–57
- Picart P, Wiermans L, Pérez-Sánchez M, Grande PM, Schallmey A, Domínguez de María P (2016) Assessing lignin types to screen novel biomass-degrading microbial strains synthetic lignin as useful carbon source. ACS Sustainabl Chem & Engineer 4:651–655
- Qu P, Huang H, Zhao Y, Wu G (2017) Physicochemical changes in rice straw after composting and its effect on rice-straw‐based composites. J of Appl Polym Sci 134:734–745
- Ravi K, García-Hidalgo J, Gorwa-Grauslund MF, Lidén G (2017) Conversion of lignin model compounds by Pseudomonas putida KT2440 and isolates from compost. Appl Microbiol and Biotechnol 101:5059–5070
- Reginatto V, Antônio RV (2015) Fermentative hydrogen production from agroindustrial lignocellulosic substrates. Brazil J of Microbiol 46:323–335
- Reilly M, Dinsdale R, Guwy A (2014) Mesophilic biohydrogen production from calcium hydroxide treated wheat straw. Intl J of Hydrog Energ 39:16891–16901
- Rodríguez-Valderrama S, Escamilla-Alvarado C, Rivas-García P, Magnin JP, Alcalá-Rodríguez M, García-Reyes RB (2020) Biorefinery concept comprising acid hydrolysis, dark fermentation, and anaerobic digestion for co-processing of fruit and vegetable wastes and corn stover. Environ Sci and Pollut Res 27:28585–28596
- Roy S, Das D (2016) Biohythane production from organic wastes: present state of art. Environ Sci and Pollut Res 23:9391–9410
- Aimen S, Ali TS, Hafiz U, Shah, Tabassum (2020) Fermentation of simple and complex substrates to biohydrogen using pure Bacillus cereus strains. Environ Technol & Innovat 4:698–704
- Shah A, Favaro L, Alibardi L, Cagnin L, Sandon A, Cossu R, Casella S, Basaglia M (2016a) Bacillus sp. strains to produce bio-hydrogen from the organic fraction of municipal solid waste. Appl Energ 176:116–124
- Shah M (2014) Isolation and screening of dye decolorizing bacteria. J of Appl & Environ Microbiol 2:244–248
- Shah TA, Shehbaz A, Asifa A, Tabassum R (2018a) Simultaneous pretreatment and biohydrogen production from wheat straw by newly isolated ligninolytic Bacillus sp. strains with two-stage batch fermentation system. BioEnerg Res 11:835–849
- Shah TA, Asifa A, Shehbaz A, Rahim U, Romana T (2017) A review on biohydrogen as a prospective renewable energy. Intl J of Biosci 11:106–130
- Shah A, Favaro L, Alibardi L, Cagnin L, Sandon A, Cossu R, Casella S, Basaglia M (2016b) Bacillus sp. strains to produce bio-hydrogen from the organic fraction of municipal solid waste. Appl Energ 176:116–124
- Shah TA, Lee C, Orts WJ, Romana T (2018a) Biological pretreatment of rice straw by ligninolytic Bacillus sp. strains for enhancing biogas production. Environ Prog & Sustainabl Energ 38:e13036
- Shah TA, Lee C, Orts WJ, Romana T (2018b) Biological pretreatment of rice straw by ligninolytic Bacillus sp. strains for enhancing biogas production. Environ Prog & Sustainabl Energ 38:e13036
- Shah TA, Raheem U, Asifa A, Romana T (2018) Effect of alkalis pretreatment on lignocellulosic waste biomass for biogas production. Intl J of Renewabl Energ Res (IJRER) 8:1318–1326
- Shah TA, Raheem U (2019) Pretreatment of wheat straw with ligninolytic fungi for increased biogas productivity. Intl J of Environ Sci and Technol 16(11):7497–7508
- Shah TA, Romana T (2018) Enhancing biogas production from lime soaked corn cob residue. Intl J of Renewabl Energ Res (IJRER) 8:761–766
- Shehbaz TAS, Asifa, Romana T (2018) Exploring lignocellulosic biomass for bio-methane potential by anaerobic digestion and its economic feasibility. Energ & Environ 29:742–751
- Shin HS, Youn JH, Kim SH (2004) Hydrogen production from food waste in anaerobic mesophilic and thermophilic acidogenesis. Intl J of Hydrog Energ 29:1355–1363
- Sluiter A, Sluiter J, Wolfrum EJ (2013) Methods for biomass compositional analysis. National Renewable Energy Laboratory (NREL), Golden, CO (protocols)
- Verhaart MR, Bielen AA, Oost J, Stams AJ, Kengen SW (2010) Hydrogen production by hyperthermophilic and extremely thermophilic bacteria and archaea mechanisms for reductant disposal. Environ Technolog 31:993–1003
- Winter CJ (2005) Into the hydrogen energy economy milestones. Intl J of Hydrog Energ 30:681–685
- Wong MH, Ok YS, Naidu R (2016) Biological waste as resource, with a focus on food waste.Environ Sci and Pollut Res7071–7073
- Xing Y, Ma H, Fan Y, Hou H, Chen J (2009) Cellulose-hydrogen production from corn stalk biomass by anaerobic fermentation. Chines Sci Bullet 54:1434–1441
- Xiong W, Reyes LH, Michener WE, Maness PC, Chou KJ (2018) Engineering cellulolytic bacterium Clostridium thermocellum to co-ferment cellulose‐and hemicellulose‐derived sugars simultaneously. Biotechnol and Bioengineer 115:1755–1763
- Yokoi H, Tokushige T, Hirose J, Hayashi S, Takasaki Y (1998) H2 production from starch by a mixed culture of Clostridium butyricum and Enterobacter aerogenes. Biotechnol Letter 20:143–147
- Zeng J, Singh D, Chen S (2011) Biological pretreatment of wheat straw by Phanerochaete chrysosporium supplemented with inorganic salts. Bioresour Technol 102:3206–3214
- Graphicalabstract.png
Graphical Design of the Experiments for bio-H2 production
{kind=link}