Study design and population. A cross-sectional study was conducted at the Immunology and Infectology Research Unit of Hospital de Infectología, Centro Médico Nacional La Raza (CMNR) del Instituto Mexicano del Seguro Social (IMSS), Mexico City, Mexico. One Hundred HIV-positive ART-naive adult PLWH (over 18 years) were recruited between 2016 and 2019. This study was performed in accordance with guidelines of the Helsinki Declaration and was approved by the Bioethical Committee of the IMSS National Research (CNIC-R-2017-785-106). Written informed consent was obtained from all participants before blood sample donation and data were processed using unique identifiers to ensure confidentiality. Baseline and follow-up clinical data, including CD4 cell count, plasma viral load (pVL), HIV diagnosis date, ART initiation date, first-line ART regimen, changes in the initial treatment regimen, time between HIV diagnosis and ART initiation were obtained from the individuals’ medical records. Demographic characteristics, including gender, age and risk factors for HIV infection were obtained by means of a validated questionnaire at the time of blood-sample donation.
Blood collection, and RNA extraction. Approximately 4 ml of peripheral blood were collected from all PLWH in ethylenediaminetetraacetic acid (EDTA) Vacutainer tubes (BD, San Jose, CA), and immediately processed to separate plasma by centrifugation at 2,500 rpm for 10 minutes at room temperature. Plasma samples were apportioned into 1.0 mL aliquots and stored at -70°C until use. For RNA extraction, a 1mL plasma aliquot was thawed per subject and centrifuged at 14,000 rpm at 4°C for 2 hours to concentrate HIV particles. RNA was extracted using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol.
Complementary DNA synthesis (cDNA), and polymerase chain reaction (PCR). The coding region of HIV-1 Gag and Protease (HXB2 nucleotides 790 to 2550) was amplified using primers and conditions as published elsewhere (23)(24). Briefly, extracted RNA was subjected to a one-step RT-PCR followed by a nested amplification. Primer sequences, reagents and cycling conditions are provided in (Additional file 1). Amplicons were visualized by electrophoresis on a 1% agarose gel stained with ethidium bromide under ultraviolet light to confirm expected amplicon size (1998 pb).
Library construction and sequencing of HIV-1 Gag and Protease. Amplicons were purified using the MinElute PCR purification kit (Qiagen, Hilden, Germany) and quantitated using Qubit ds DNA Assay Kit (ThermoFisher, Waltham, MA, USA) according to the manufacturers' instructions. DNA libraries were prepared using the Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA), using 1 ng of input DNA, and indexed using Nextera XT Index Kit (Illumina, San Diego, CA, USA), according to manufacturers’ instructions. Libraries were checked for size and molarity using the High sensitivity D1000 ScreenTape (Agilent, Santa Clara, CA, USA) on a 4200 TapeStation (Agilent, Santa Clara, CA, USA). DNA libraries were pooled at 4nM and further diluted to 9pM. Libraries were sequenced on a MiSeq platform (Illumina, Santa Clara, CA, USA) using 2x250 cycles (MiSeq Reagent Kits v.2) with 5% PhiX as a control.
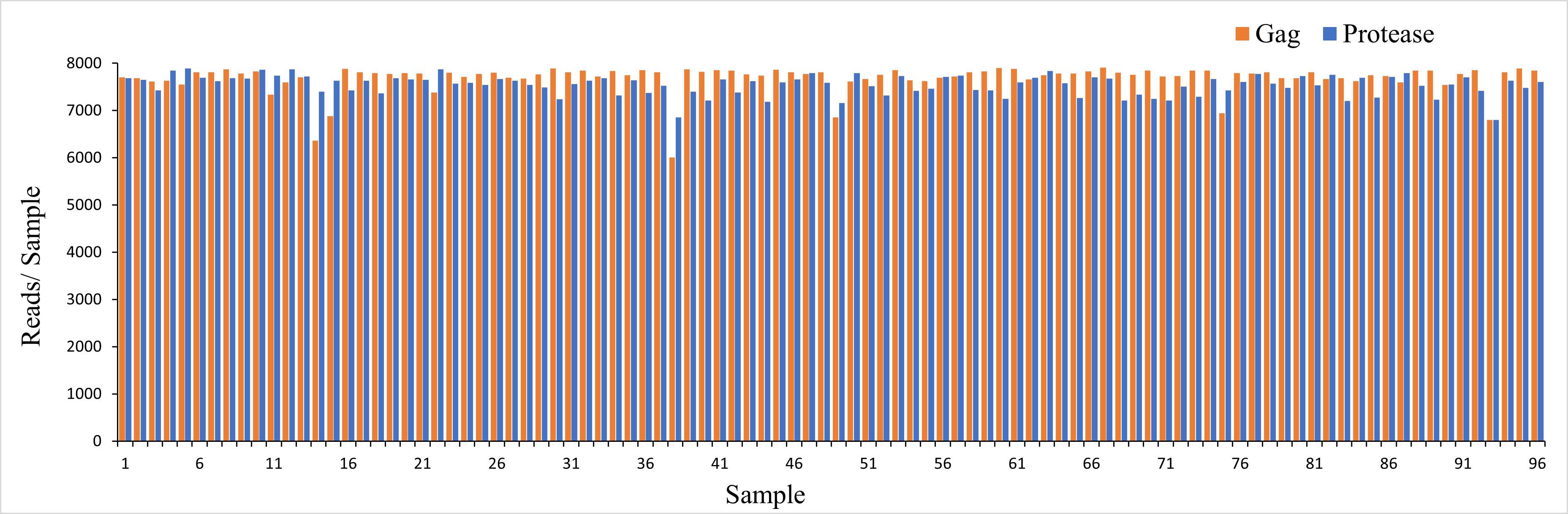
Sequence data analysis. In order to identify HIV Gag and Protease DRMs we took a three-fold approach, first we assembled full length sequences using automated Target Restricted Assembly Method (a TRAM, version 2.1.0) locus assembler, an iterative assembler that performs reference-guided local de novo assemblies using a variety of available methods (25), with Trinity (v2.9.0) (26) as the de novo assembler and the HIV-1 HXB2 sequence (GenBank: K03455) as the template. We also determined codon-specific variants as well as their relative frequencies using an alignment and variant-calling pipeline for Illumina deep sequencing of HIV-1, based on the probabilistic aligner profile Hidden Markov Model for HIV (HIVMMER, version 0.1.2) (27) with the HXB2 as reference sequence. Finally, we aligned the full length sequences to the HXB2 reference using Burrows-Wheeler Aligner (BWA, version 0.7.17), a software package for mapping low-divergent sequences against a large reference genome (28). Phylogenetic trees were generated from sequences obtained from 96 treatment-naive Mexican PLWH. We identified two viral sequences originating from individual 37, located in different branches. HIV reference strains representing all HIV subtypes were used (Additional file 2). The nucleotide sequences were aligned using the multiple sequence alignment program Clustal-omega (version 1.2.1) (29) and edited manually using Seaview (version 5.0.4), a multiplatform program designed to facilitate multiple alignment and phylogenetic tree building from molecular sequence data through the use of a graphical user interface (30). A neighbor joining distance tree was constructed for each region separately: gag (1683 nucleotides) and protease (303 nucleotides). In order to provide statistical support to the constructed trees, they were subjected to 10,000 bootstrap replicas. Trees were custom-drawn using the ETE toolkit version 3 (31). To ensure accuracy, a detection threshold of 1% was used (based on the intrinsic error rate of the system (8)). To calculate sequencing depth we used Sequence Alignment/Map (SAM tools, version 1.10) (32). We achieved high level depth, with a median sequencing of 7616.84 reads (interquartile range (IQR), 7543.73 -7793.41) for Gag and Protease respectively (Additional file 3).
Drug resistance mutations (DRMs). HIV Drug Resistance Database (HIVDB; version 8.8) and the International AIDS Society 2019 drug resistance mutations in HIV-1 list were used (33)(34). Gag sequences were analyzed for the presence of specific mutations previously reported to be associated with resistance to PI and treatment failure; these were: E12K, V35I, E40K, G62R, L75R, R76K, Y79F, T81A K112E, G123E, M200I, H219Q/P, Q369H, V370A/M, I389T, V390A/D, R409K, E468K, Q474L, P497L; as well as mutations in Gag CS: V128I/T/A, Y132F, A360V, V362I, L363M/F/C/N/Y, S368C/N, S373P/Q, A374P/S, T375N/S, I376V, G381S, E428G, Q430R, A431V, K436E/R I437T/V, L449F/P/V, S451T/G/R, P452S/K and P453A/L/T (20)(35). Different detection thresholds (1%, 2%, 5%, 10% and, 20%) were used to identify the prevalence of low abundance (or minority) mutations as these could be missed using standard clinically relevant threshold (20%, Sanger sequencing).
{kind=link}
{kind=link}