The TpDPP COF nano-spheres were synthesized by the reaction between the 0.03 mmol triformylphloroglucinol (Tp), and 0.045 mmol 3,8-diamino-6-phenylphenathridine (DPP) in the presence of a catalytic amount (10-15mL) of trifluoroacetic acid (figure 1a and section S-2). The reaction conditions first led to the direct nucleation of the organic building blocks, followed by spatiotemporal growth. The formation of the organic framework structure of the COF nano-spheres could be ascertained by the intense PXRD peak at [3.65±0.06] (2θ) imputed to their (100) plane diffractions (Figure 1d).29 The experimental diffraction pattern manifests good agreement with the slipped-AA stacking models¢ simulated powder diffraction pattern. Additionally, the Pawley refinement between the simulated slipped-AA model and the experimental powder diffraction pattern using the Reflex Plus module of the Material Studio (Figure S2) showed good agreement (Rp = 4.5%, Rwp = 5.4%). The N2 adsorption analysis revealed a surface area (SBET) of 686 m2g-1 with a uniform pore size (~1.9 nm) distribution (Figure S8 and S9). The FTIR spectra of TpDPP nano-spheres displayed intense peaks at 1600–1625 (–C=O), 1579 (–C=C), 1277 cm-1 (–C–N), and peaks at 1671 (–C=O of free –CHO), 2851-2920 (–C–H of free –CHO), 3067-3352 cm-1 (–N–H stretching of free –NH2) (Figure S5). The FTIR spectra suggested the formation of a β-ketoenamine backbone30-32 and the presence of free aldehyde and amine functionalities on the sphere surface. The presence of free aldehyde and amine functionalities indicates incomplete crystallization and structural defects. The zeta potential of the colloidal COF nano-spheres in DCM was measured to be +65.4 mV at 20 °C, which suggests the presence of the defused positive charges at the interface between the solid COF nano-spheres and the solvent molecules (Figure 2e). As expected, the dispersible COF nano-spheres' zeta potential dropped to +9.5 mV after neutralization with triethylamine (Et3N) (Figure 2f). The dynamic light scattering (DLS) analyses revealed the as-synthesized COF nano-spheres¢ average size to be in the range of 590-610 nm (Figure 2e-f). The size and zeta potential of COF nano-spheres remained unaltered (626 nm and +65.4 mV as-synthesized vs. 632 nm and +61.7 mV) for four months (Figure 2g and 2j). Such a constant presence of permanent positive charges at the solid-liquid interface made these nano-spheres stable in solution and prevented their aggregation.
The high surface area and defects within the COF nano-spheres made them a promising candidate for the physisorption of catalytically active (Et4N)2[FeIII(Cl)bTAML] molecules within the condensed space. In enzyme catalysis, the amide functionalities of the enzyme backbone or the charged amino acid side chains can influence the active sites' electronic properties through long-range interactions.33 We envisaged that the hydrogen bonding between the first coordination sphere amide functionality (–C=O) of the (Et4N)2[FeIII(Cl)bTAML] molecules and the free –NH2 of the COF nano-spheres has a significant effect on the catalytic reactivity and selectivity. Such increased reactivity towards oxidation reactions upon binding Lewis acids to the ligand carbonyl in the related oxo Mn(V)-TAML complexes has been reported earlier.34,35 The Raman spectra of the (Et4N)2[FeIII(Cl)bTAML] catalyst obtained upon 785 nm excitation revealed the presence of an Am I (–C=O stretch) band at 1616 cm-1 and an Am II (C–N stretch) band at 1569 cm-1. The catalyst Am I (–C=O stretch) band red-shifted to 1601 cm-1, and a new peak appeared at 1621 cm-1 due to the immobilization of the catalyst inside the COF nano-spheres. Similarly, the catalyst Am II (C–N stretch) band blue-shifted to 1573 cm-1, and a new peak appeared at 1562 cm-1 (Figure S31). This shift in –C=O and C–N stretch bands indicates the presence of N─H···O hydrogen bonding between the free amine of the COF nano-sphere and the amide carbonyl of the immobilized catalyst.36,37 The XPS analysis of the (Et4N)2[FeIII(Cl)bTAML] catalyst revealed the deconvoluted C 1s spectra, which indicates the presence of two distinct peaks at 286.1 and 287.1 eV corresponding to the amide carbon and the diamide carbon, respectively (Figure S32-33). While immobilized inside the COF nano-sphere, these two peaks appeared at almost similar regions (286.6-286.9 eV) due to the hydrogen bonding and could not be appropriately resolved.38
1.0 mg of solid COF nano-spheres can immobilize ~95% (Et4N)2[FeIII(Cl)bTAML] catalyst from 1 ml 0.2 and 0.4 mM solution in acetonitrile (Figure S14-15). We believe that the positively charged COF nano-sphere matrix, containing –NH3+ and N–H functional groups, drives the immobilization of negatively charged [FeIII(Cl)bTAML]2- molecules. Consequently, we can control the wt% loading of the immobilized catalyst within the dispersible COF nano-spheres by increasing the amount of COF nano-spheres and keeping the concentration of the catalytic solution (1 ml 0.4 mM solution in acetonitrile) constant (Figure S55 and section S-19). We deliberately formulated COF nano-spheres for catalyst immobilization because of their adsorption capacity, by considering a similar quantity of highly crystalline COF powder39 and films (Figure S12). TpDPP COF powder (1565 m2g-1) and thin-film (1265 m2g-1) do have a higher surface area compared to the nano-spheres (686 m2g-1). However, the catalyst adsorption capacity of the COF nano-spheres (94.8%) is 2-3 times higher than the COF powder (47%) and COF thin-films (21%). Furthermore, the COF nano-spheres' ability to transmute into thin-films gave us another avenue to fabricate COF thin-films with a higher amount of immobilized catalyst. The dispersible COF nano-spheres achieved 90% of their absorption efficiency within 90 seconds. The maximum uptake of the (Et4N)2[FeIII(Cl)bTAML] catalyst by the as-synthesized TpDPP COF nano-spheres was found to be 378.9 mg g-1. We have performed the identical absorption experiment with another iron-based electrophilic homogeneous catalyst [FeII(S, S-PDP)](SbF6)2, which is a cationic complex and also known to catalyze the hydroxylation reaction of C-H bonds (Figure S16-17). The immobilization of [FeII(S,S-PDP)](SbF6)2 into the porous COF nano-sphere matrix was only 36%, due to the ionic repulsion between the two cationic species.
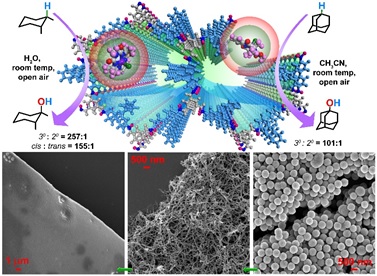
In CH3CN, [FeIII(Cl)bTAML]2-@1X TpDPP COF nano-spheres (catalyst immobilized from 1 ml 0.4 mM acetonitrile solution by 1 mg of COF nano-spheres) was found to catalyze the hydroxylation of adamantane (8 mM; 20 eq.) with NaOCl (0.1 M, 250 eq.) to yield 1-adamantol as the primary product with a 45:1 selectivity for the 3° C-H bond over the 2° C-H bond (Figure 3c). Optimization reactions performed with various (Et4N)2[FeIII(Cl)bTAML] loaded COF nano-spheres indicated that the maximum reactivity (73% conversion, 70% yield) and selectivity (3°:2º=101:1) was observed for [FeIII(Cl)bTAML]2-@2X TpDPP COF nano-spheres (catalyst immobilized from 1 ml0.4 mM acetonitrile solution by 2 mg of COF nano-spheres) (Figure S55). This optimization study indicated that maximum loading of COF nano-spheres with (Et4N)2[FeIII(Cl)bTAML] reduced the selectivity of oxidation due to possible processes involving dimeric oxoiron intermediates formed due to their proximity. On the contrary, when the loading was significantly reduced, the conversions decreased, indicating that the rates slowed due to excess COF present. Based on this data, [FeIII(Cl)bTAML]2-@2X TpDPP COF nano-spheres were employed as the catalyst for all subsequent reactions. Control reactions
performed with only NaOCl (0.1 mM) or bare COF nano-spheres afforded <5% products. We have explored the oxidation of a series of substrates containing unactivated 3° C-H bonds and 2° benzylic C-H bonds using [FeIII(Cl)bTAML]2-@2X TpDPP COF nano-spheres as the catalyst and NaOCl as the oxidant in CH3CN under the optimized conditions (Figure 3f and section S-19). The heterogeneous oxidation of adamantane, having twelve 2° C-H and four 3° C-H bonds afforded selectively 1-adamantanol in ~70% yield with remarkably high regioselectivity (i.e., 101:1) of the 3° C-H bond over the 2° C-H bond (Figure 3c). In the case of cis-1,2-dimethylcyclohexane, trans-1,2-dimethylcyclohexane, and cis-decalin (Figure 3f), mainly 3° hydroxylated products were obtained in 90%, 42%, and 85% yields with 99% stereo-retention. Substrates having activated benzylic C-H bonds such as ethylbenzene (EB), diphenylmethane (DPM), dihydroanthracene (DHA), and xanthene were explored. Primarily ketone products were formed for all these substrates with high conversion and yield (Fig. 3f-h). The oxidation of 3º C–H bonds in simple hydrocarbons was then extended to natural product derivatives. For example, cedryl acetate, a natural product derivative of cedrol found in essential oil, having a rigid structure with five 3° C–H bonds, affords a single hydroxylated product in 70% yield (Figure 3h). Ambroxide, a naturally occurring terpenoid used in perfumery, undergoes exclusive oxidation at the alpha ethereal C–H bond predominantly among many other electronically and sterically accessible secondary and tertiary C–H bonds (Figure 3h). Finally, the use of alkenes as substrates led to the corresponding epoxides' formation in high yields (60-99%) (Figure 3g). During these catalytic reactions, the products formed parallel those that have been found under homogeneous conditions using Fe-bTAML analogs and NaOCl. We have previously demonstrated40 that (Et4N)2[FeIII(Cl)bTAML] reacts with NaOCl to form the active oxoiron(V) intermediate, which is responsible for substrate oxidation. For the [FeIII(Cl)bTAML]2-@2X TpDPP COF nano-spheres, X-band EPR spectrum at 123K (Figure S34) manifested a rhombic S= ½ species with g = 2.00, 1.93, and 1.73, which indicated that the same oxoiron(V) intermediate was generated inside the COF nano-spheres, which confirms that the overall mechanism of oxidation remained unchanged upon immobilization of the catalyst.
Although the overall mechanism of oxidation remains unchanged, several aspects are worth noting. The selectivity obtained for the oxidation of hydrocarbons in the [FeIII(Cl)bTAML]2-@COF nano-spheres far exceeds what is observed under homogeneous conditions. For catalytic oxidation of adamantane using (Et4N)2[FeIII(Cl)bTAML]/NaOCl under homogeneous conditions in air, the selectivity towards 3º C-H is reduced nearly four-folds (110:1 in the absence of air to 31:1 in the presence of air). This reduction in selectivity occurs since some of the carbon-centered radicals formed during C-H abstraction react with O2 via the "non-rebound" pathway leading to an additional free radical auto-oxidation pathway. In fact, during the homogeneous oxidation of adamantane in air, 1-adamantanol is formed along with other products such as 2-adamantanol and 2-adamantanone (9-10%), which significantly reduces the regioselectivity of 3° C-H bond oxidation over 2° C-H bonds.
However, the regioselectivity obtained for adamantane oxidation by [FeIII(Cl)bTAML]2-@2X TpDPP COF nano-spheres, in the presence of O2, is similar (3°:2° = 101:1 based on the statistical correction for the number of C−H bonds) to that obtained for reactions that were performed under homogeneous conditions with (Et4N)2[FeIII(Cl)bTAML] in the absence of O2.41,42 Such regioselectivity could be due to the total shutdown of the free-radical pathway since the hydrophobic scaffold of the COF nano-spheres creates an appropriate binding pocket for both the anionic macrocyclic catalyst (via ionic interactions) and the substrate (via predominantly hydrophobic interactions). Consequently, the carbon-centered radical formed upon C-H abstraction does not escape to the bulk to react with O2, thereby initiating a free-radical process that would significantly reduce the selectivity. Therefore, the oxidation mechanism solely involves the "rebound" of the carbon-centered radical with the Fe(IV)-OH to form the hydroxylated product with high regioselectivity and stereo-retention.43 Additionally, the prototype Fe-bTAML complex under homogeneous conditions displays poor turnover numbers for catalytic oxidation of hydrocarbons since it undergoes acid or base induced demetallation with chemical oxidants (e.g., mCPBA or NaOCl) (pH=12.6) (Figure S42). We need to i) modify the head part of the Fe-bTAML by introducing a –NO2 group to increase its robustness to hydrolytic degradation,44,45 and ii) use sodium phosphate buffer to maintain the pH of the reaction to achieve a high turnover number. In this case, high catalytic efficiency was observed with the prototype Fe-bTAML encapsulated inside the COF nano-spheres. Besides, no further use of base was required for maintaining the pH. The [FeIII(Cl)bTAML]2-@2X TpDPP COF nano-spheres could be recycled up to 4 cycles without altering the activity and selectivity of the catalyst (Figure 3d). The improved reactivity of (Et4N)2[FeIII(Cl)bTAML], when incarcerated inside the COF nano-spheres, resembles enzymes like cytochrome P450 in which the hydrophobic cavity facilitates oxidation of water-insoluble hydrophobic substrates by efficiently partitioning them inside the cavity. We wanted to explore if the oxidation of water-insoluble substrates could be carried out with [FeIII(Cl)bTAML]2-@2X TpDPP COF nano-spheres using water as the solvent. It should be noted that homogeneous C–H functionalization in water using Fe-complexes is an arduous task since the high-valent oxoiron intermediates formed upon addition of oxidant react with the O-H bond of water, which is present in much greater concentration. We have earlier shown the in 100% water, (Et4N)2[FeIII(Cl)bTAML] catalyzes water oxidation to form O2 via the oxoiron(V) intermediate. However, our attempts for C–H functionalization were unsuccessful, as the COF nano-spheres aggregated in water.
Hence, we decided to construct TpDPP COF thin-films from the catalyst immobilized TpDPP COF nano-spheres via covalent self-assembly. Individual TpDPP COF nano-spheres undergo covalent self-assembly in the water-DCM bilayer or even while drop-casted on top of any support, and eventually form uniform COF nano-films. The COF nano-films are more crystalline and porous than COF nano-spheres and do not undergo any further transmutation in water. The covalent self-assembly driven by the free amine and aldehyde functionality of the COF nano-spheres occurs via a unique, dynamic process previously unheard of. The attractive capillary forces and convective transport of the COF nano-spheres drive their self-assembly. Consequently, when two separate spheres come in contact, they start reacting via a reversible Schiff base reaction, which initiates the fibers/threads' protrusion at their interface (Figure 3a-i and section S-2). The distribution of fibers enhances with time to accomplish the formation of a COF nano-film of thicknesses of 250-270 nm after three days (Figure 5e-f and S44). It is quite fascinating that the probability/chance of immobilized (Et4N)2[FeIII(Cl)bTAML] molecules escaping from the COF nano-spheres to the bulk during the covalent self-assembly was nullified entirely by the non-covalent interactions of the catalyst molecules with the COF binding pockets. The dynamic transmutation from the nano-spheres ® nano-fibers ® nano-films did not affect the immobilized catalyst molecules and culminated in highly fecund catalytical COF thin-films. The EDX analysis confirmed uniform distribution of (Et4N)2[FeIII(Cl)bTAML] within COF nano-spheres, nano-films, and every intermediate phase during this transmutation process (Figure S37). We have monitored the spheres ® fibers ® film transmutation with confocal imaging to ascertain the immobilized catalysts' enduring stability during the covalent self-assembly (Figure 4j-m and section S-15). The confocal imaging revealed the uniform adsorption and distribution of (Rhodamine B) dye molecules inside the COF nano-spheres and COF fibers, which manifested their high porosity and adsorption capacity. These (Et4N)2[FeIII(Cl)bTAML] molecules are confined within the hydrophobic pockets of the COF thin-film. Because of the hydrophobicity of the [FeIII(Cl)bTAML]2-@TpDPP COF thin-films (contact angle 123.2°), only the hydrophobic substrates could contact the active catalytic sites but not the water molecules (Figure 5c and S41). The wt% loading of the (Et4N)2[FeIII(Cl)bTAML] catalyst within the COF nano-spheres and the COF thin-films were found to be 23 wt% and 19 wt%, respectively (Section S-7). The ICP-MS analysis of the catalyst immobilized COF nano-spheres and COF film showcased the percentage of metal (Fe) loading to be 4.5 and 3.6 wt% respectively.
We could achieve the heterogenous C–H functionalization in water for the very first-time using (Et4N)2[FeIII(Cl)bTAML]- immobilized COF nano-films. To a dispersion of [FeIII(Cl)bTAML]2-@TpDPP COF thin-film in water was added adamantane and NaOCl (0.1 M, 250 eq. added iteratively), and the reaction was allowed to proceed for 1.5 hrs. Analysis of the products showed the formation of 1-adamantanol with 45% yield (47% conversion) and 141:1 (3°:2°) selectivity Figure 5a). With cis-dimethyl cyclohexane as the substrate, cis dimethyl cyclohexanol (yield 75%) was formed as the predominant product with 257:1 (3°:2°) regioselectivity and 155:1 (cis:trans) stereoretention from cis-dimethyl cyclohexane (Figure 5a). After the reaction, the [FeIII(Cl)bTAML]2-@TpDPP COF thin-film was recovered easily by centrifugation. The same thin-film was used for four more subsequent reactions, and the yield and selectivity were unchanged even after the fourth cycle (Figure 5d). We believe that the cis-dimethylcyclohexane preferentially partitions inside the COF nano-spheres in the COF film, and this allows the reaction of water-insoluble substrate oxidation in water. We could not achieve more than 65% conversion in each cycle, probably due to the substrate's similar binding affinity and the product inside the COF film in water. C–H bonds of other substrates such as cis-decahydroanthracene, ethylbenzene (EB), diphenylmethane (DPM), and ambroxide were also oxidized using the [FeIII(Cl)bTAML]2-@TpDPP COF thin-film/NaOCl in water with high catalytic yield and excellent selectivity (Figure 5a-b and section S-19). Under optimized conditions, for cis-decahydroanthracene, 3° hydroxylated products were obtained in 83% yield with 99% stereoretention without the formation of any 2° hydroxylated products. Only keto products were found for the functionalization of EB, DPM, and ambroxide with 99% (acetophenone), 99% (benzophenone), and 91% (sclareolide) yields, respectively (Figure 5a-b and section S-19).
We performed a proof-of-concept flow catalysis46,47 using catalyst immobilized COF thin-films on a macroporous solid polymeric support to establish the generality of this C–H functionalization approach (Figure 5g and section S-18,19). 90 mg (0.66 millimoles) adamantane was dissolved in 25 ml CH3CN (0.66 millimoles of bromobenzene as internal standard). It was passed through ~5 mg of [FeIII(Cl)bTAML]2-@TpDPP COF thin-film (19wt% catalyst loading) for several cycles with iterative addition of NaOCl (0.06 M). After the 60th cycle, this inflow catalysis showcased the 72% catalytic yield of 1-adamantanol with an excellent turnover number of 355±5 (Figure 5h).
{kind=link}