Chemicals and antibodies
The following chemicals were used: recombinant human prorenin (ab93266) was from Abcam (Cambridge, MA) and SKF10047 (1079) was purchased from Tocris Bioscience (Bristol, UK). Artificial cerebrospinal fluid (aCSF) (CZ0530) was purchased from Leagene (Leagene Biotechnology, Beijing, China). ATP Assay Kit (S0026), Mito-Tracker Green (C1048), ER-Tracker Red (C1041) were purchased from Beyotime (Shanghai, China). The following antibodies were used: rabbit anti-sigma-1R (#15168-1-AP) were from Proteintech; sheep monoclonal antiserum against prorenin/renin (GTX79677, Gene Tex, San Antonio, TX); rabbit anti-CD86 (#91882), anti-CD206 (#91992), anti-MFN2 (#9482), anti-OPA1 (#67589), anti-Drp1 (#8570), anti-GAPDH (#5174), anti-GRP75 (#3539), anti-ASC (#13833), anti-Caspase-1 (D7F10) and IL-1β (D3U3E) were from Cell Signalling Technology (Beverly, MA); mouse anti-IP3R (#SC377518) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit monoclonal antiserum against TTC11/Fis1 (ab156865), mouse monoclonal to β-actin (ab8226), VDAC1 (#ab14734 1:5000 for WB); rabbit polyclonal antiserum against NLRP3 (ab214185) were from Abcam. Goat anti-mouse IgG H&L (Alexa Fluor 488) (ab150113); Goat anti-Rabbit IgG H&L (Alexa Fluor 594) (ab150080); Donkey Anti-Rabbit IgG H&L (Alexa Fluor 647) (ab150075); Donkey Anti-Rabbit IgG H&L (HRP) (ab6802) and Donkey Anti-Mouse IgG H&L (HRP) (ab6820) were purchased from Abcam (Cambridge, UK).
Experimental design
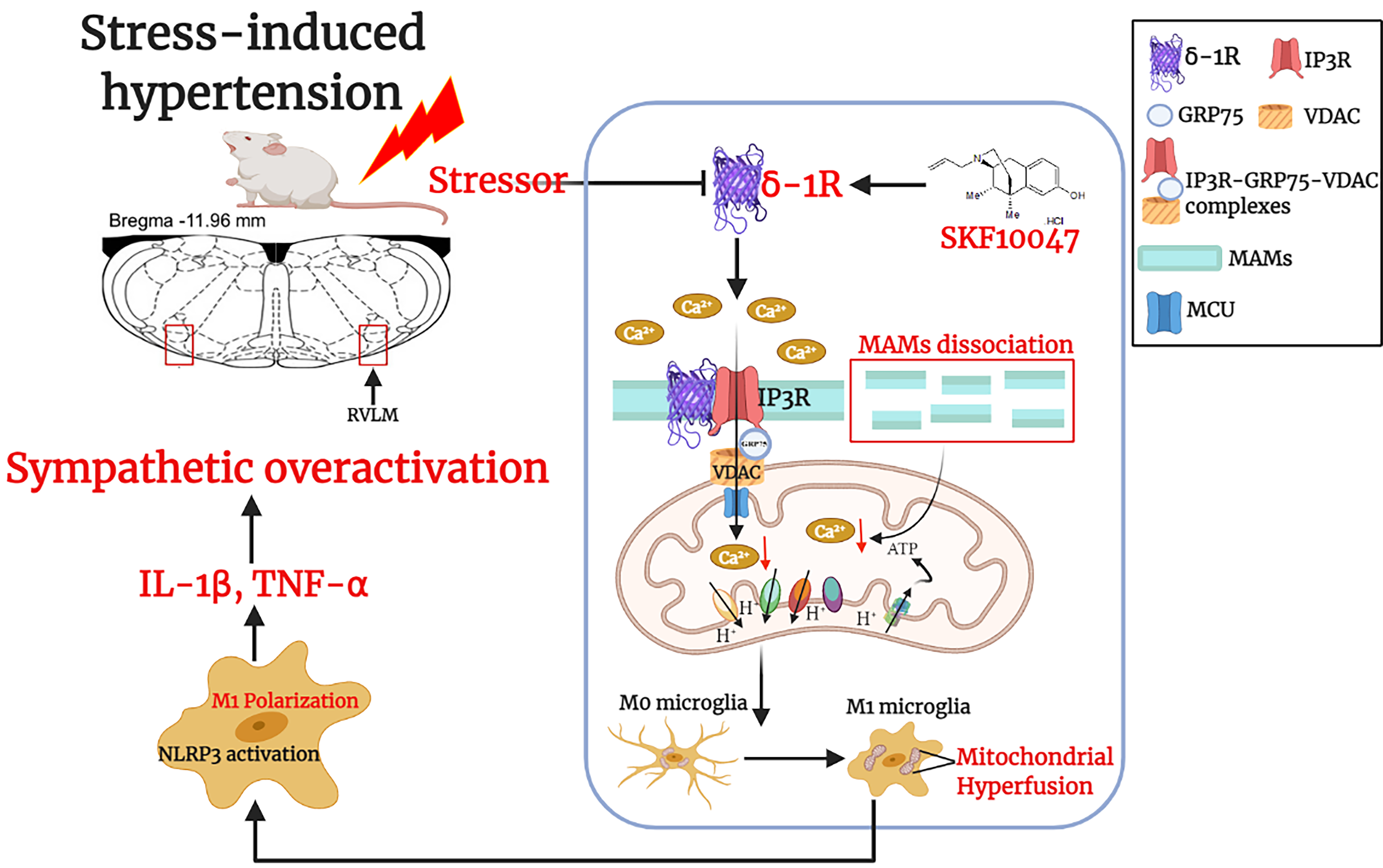
Experimental 1: To explore the expression and the regulatory roles of σ-1R in SIH in vivo, Adult Sprague-Dawley rats (male, 8 weeks old, 250–300 g) were randomly assigned to three groups (6 rats each): (i) normotensive (Ctrl); (ii) stress-induced hypertensive (SIH); (iii) SIH + SKF10047 (a prototype σ-1R agonist). The rats were housed under 12-h light/dark cycle condition in a temperature-controlled room at 24℃ with standard food and tap water ad libitum. SKF10047 was used to activate σ-1R in SIH rats, specifically, 100µM intracisternal infusion into SIH rats was carried out by osmotic minipump once daily for 1 week (from stress day 8 to day 15).
Experimental 2: To identify the roles of σ-1R in mitochondrial associated endoplasmic reticulum membranes (MAMs) as well as to investigate the antagonistic effects of σ-1R activation in pro-inflammatory phenotype (M1) switching. The primary microglial cells were obtained and cultured ex vivo. Primary microglial cells were seeded and grown for 7 days in DMEM supplemented with 10% (vol/vol) serum (basal conditions). Next, they were divided into 3 groups: (i) control (Ctrl, vehicle); (ii) prorenin (20 nM/L for 24 h) treatment (PRO) [4, 30]. (iii) PRO + SKF10047 (100µM for 24 h)[31]. The microglia-like BV2 cells in each cohort were used in the case that the primary microglia were inadequate in the above-mentioned experiments.
Animal preparation
Eight weeks old male Sprague-Dawley (SD) rats (260-300g) and 1-old-day SD rats were used for this study and purchased from the Animal Laboratory Center of Fudan University. All experiments were performed on 110 adult male SD rats and 50 1-old-day SD rats, with a mortality rate of 5% due to intolerance of brain surgery, or death during stress. We have described the groups and the design of the SIH model for this study. In short, rats were subjected to electric foot shock with noises. They were put in a Plexiglas chamber (26 cm × 21 cm × 26 cm) with a grid floor made of stainless-steel rods (0.3 cm diameter, spaced 1.0 cm apart), connected to a shock generator. A series of foot shocks, comprised of 0.5-mA intensity of 1-s duration with shock interval of 4 min, were controlled and generated electronically. Synchronously, noises (range 88–98 dB) produced by a buzzer were given as a conditioned stimulus. After acclimatization for a few times, the stressed group rats were subjected to stress for 2 h twice daily for 15 consecutive days. The control group underwent sham stress. During the days of stress, BP and HR (heart rate) in conscious rats were measured by non-invasive tail-cuff system (ALC-NIBP, Shanghai Alcott Biotech) every 4 days after treatments according to our previous study[32]. To adapt to the BP recording procedure, the animals were placed on the warm platform for 30 min. BP was measured in a proper environment (RT, lightning, and noise-free atmosphere). BP measurements were replicated for at least 3 times, and the average value was obtained.
Cell culture
As described previously [33], primary cultures of microglial cells were prepared from the medulla oblongata covering the RVLM, which was removed from 1-day-old Sprague-Dawley rats after decapitation. The RVLM was identified according to the atlas of Watson and Paxinos[34]. Both sides of the RVLM (about 1.5- to 2.5-mm lateral to the midline and medial to the spinal trigeminal tract) were collected using micropunches with a 1-mm inner diameter burr. Next, Hanks balanced salt solution dissecting medium containing glucose, bovine serum albumin (BSA), and HEPES, as well as 0.025% trypsin was used to incubate the minced tissue at 37℃ for 20 min. Then, cells were cultured at a density of 3 × 105 cells/ cm2 in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium (Biochrom, Berlin, Germany) supplemented with 10% fetal bovine serum (FCS; Biochrom), 0.1 mg/mL streptomycin and 100 U/mL penicillin in poly-L-lysine-coated 75 cm2 culture flasks. After 3 days, the culture medium was removed and replaced with fresh medium and kept at 37℃ in a humidified 95% O2/5% CO2 incubator. On day 9, the cells were re-suspended after centrifugation (150 × g for 10 min)[35]. The purity of cultured microglia was >90% when evaluated by flow cytometry.
BP measurements and RSNA recording
We recorded RSNA when the general procedures for acute experiments were ready, the RSNA recording method was adapted from our previous study[5]. In anesthetized rats (urethane 800 mg/kg and α-chloralose 40 mg/kg ip), the right formal artery was catheterized for the recording of BP and HR using a Powerlab system (AD Instruments). Next, the left renal sympathetic nerves were exposed, identified and carefully dissected free from the surrounding connective tissue, and then they were placed on a pair of silver recording electrodes (Teflon 786500, A-M Systems Inc., Sequim, WA). Both the nerves and the electrodes were covered with Kwik-Sil gel (World Precision Instruments). The nerve signal was amplified (bandwidth: 100–3,000 Hz) with a Grass P55C preamplifier (model P 18D, Grass Instruments). The amplified discharge was monitored on a storage oscilloscope (model 121 N; Tektronix, Beaverton, OR), and the maximum value of RSNA was calculated after the rat was overdosed by narcotic euthanasia (200 mg/kg). Baseline RSNA was taken as a percentage of maximum after the background noise was subtracted. The background noise level was recorded 15–20 min after the rat was euthanized using the unit conversion of the PowerLab Chart system.
Implantation of Intracisternal Osmotic Minipump
The procedures were performed as described in our previous study[5]. Briefly, the rats were anesthetized as above-mentioned. After a midline dorsal neck incision, the dura was perforated with a 22-gauge steel needle, and following cerebrospinal fluid leakage, a PE-5 catheter (Clay Adams, Sparks, MD) was advanced 5 mm into the cisterna magna and sealed to the dura with tissue glue. The outer end of the catheter was connected to a micro-osmotic minipump (Alzet 1007D, Durect Co., Cupertino, CA). The SKF10047 (100 µM) microinjection was delivered by osmotic minipump. Rats were used in normal physiological condition after the operation in subsequent experiments. After SKF10047 microinjections, rats were treated with 1000 units of penicillin through muscle injection to prevent infection.
Transmission electron microscopy
Transmission electron microscopy analysis was performed as stated in previous publications [10, 32, 36]. The cells were collected in the EP tubes after the cell medium was decanted, the cells were fixed with 2% paraformaldehyde plus 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.2-7.4, and then they were osmicated, rinsed with phosphate buffer, dehydrated, and embedded in epoxy resin, which then they were allowed to polymerize for 24 h at 70 °C. Blocks containing microglia were sectioned using an ultramicrotome (Ultracut; Leica) at 70–80 nm. Thin sections were collected on grids and stained with uranyl acetate and lead citrate. Grids were examined under a transmission electron microscope (H-700; Hitachi, Tokyo, Japan) at 80 kV.
Flow Cytometry analysis of M1/M2 phenotype
For M1 phenotype classification via flow cytometry analysis, primary microglial cells were cultured at a density of 3 × 105 cells/ cm2 and starved with DMEM/DF12 with 2% FBS overnight. As described previously [37], after being treated with prorenin (20 nM/L for 24 h) and/or SKF10047 (100µM/L for 24 h), microglial cells were harvested followed by centrifugation at 300×g for 5 min at 4 °C. Cells were surface-stained for 45 min in the darkroom with fixation and permeabilization buffer as per the manufacturer’s instructions, then they were incubated with PE-conjugated anti-mouse CD86 (0.125 μg/test; eBioscience, Waltham, MA, USA) and APC-conjugated anti-rat CD206 (10 μl/106 cells; R&D, MN, USA). Finally, the microglial cells were washed twice with PBS and were resuspended in 500 μl PBS. Data were analyzed using Flowing software v2.5.1 (FACS Calibur running CellQuest Pro; Becton Dickinson, UK) and the percentage of positive cells was documented.
Immunofluorescent Staining and Confocal Microscopic Imaging
As stated in previous literature[32], immunofluorescent staining was detected by laser confocal microscopy. To determine the co-localization of pro-inflammatory phenotype (M1) microglia CD86 with σ-1R, MFN2, OPA1, and NLRP3, the rats were euthanized with excessive pentobarbital sodium (200 mg/kg, ip), then 0.9% NaCl solution and 4% paraformaldehyde in 0.1M phosphate buffer solution (PBS) were used to perfuse the aorta through a constant flow pump. RVLM sections were collected and post-fixed with 4% paraformaldehyde for 4 h. The RVLM sections were then dehydrated in 20%-30% sucrose until they sank to the bottom of the solution, respectively. Free-floating 30 μm coronal sections were cut on a cryostat (Microm, Germany) and were frozen at −20°C floated in 0.01M PBS (pH 7.4). These coronal sections containing the RVLM were mounted on slides after 3-5 min washes and then incubated with 0.3% Triton X-100 for 30 min followed incubation by 5% horse serum for 1 h at 37 °C to block non-specific protein. RVLM coronal sections were incubated with primary antibody overnight at 4 °C. The Goat anti-mouse IgG H&L (Alexa Fluor 488) (ab150113); Goat anti-Rabbit IgG H&L (Alexa Fluor 594) (ab150080); Donkey Anti-Rabbit IgG H&L (Alexa Fluor 647) (ab150075) secondary antiserum were used as secondary antibody. σ-1R, MFN2, OPA1, prorenin, and NLRP3 colocalization with M1 phenotype microglial marker CD86 was investigated in vivo. For in vitro experiment, Primary microglial cells were cultured on glass cover slips and treated with prorenin and /or SKF10047 for 24h. After being washed with PBS, cells were fixed with 4% PFA for 10 min, then incubated with blocking buffer (5% BSA and 0.1% Triton X-100; Gentihold, Beijing, China) for 30 min, primary antibodies (σ-1R, 1:400) overnight at 4 °C. Subsequently incubated with correspond fluorochrome-conjugated secondary antibodies at room temperature for 1 h. immunoreactivity manifested as specific green or red fluorescence. Images were captured using confocal microscope (LSM800, Carl Zeiss Microscopy Ltd, Cambridge, MA).
Western blot analysis
Microglia isolated from the RVLM tissue of each mouse were homogenized in lysis buffer with 1% NP40, 1 mM PMSF. In brief, equivalent amounts of protein (20 μg per lane) were loaded and separated by SDS/PAGE in 8%-15% gradient gels and were transferred to PVDF membrane (Invitrogen, Carlsbad, CA, USA). The membranes were blocked with 5% nonfat milk solution in tris-buffered saline with 0.1% Triton X-100 (TBST, Invitrogen, Carlsbad, CA, USA) for 1 h, and then incubated overnight at 4 ℃ with the primary σ-1R antibody (1:1000), CD86 antibody ( 1:1000), MFN2 antibody (1:1000), Fis1 antibody (1:1000), Drp1 antibody (1:1000), OPA1 antibody (1:1000), IP3R antibody (1:1000), GRP75 antibody (1:1000), VDAC antibody (1:1000), NLRP3 antibody (1:1000), ASC antibody (1:1000), Caspase-1 antibody (1:1000), IL-1β antibody (1:1000), GAPDH antibody (1:1000) and β-actin (1:1000) antibody dilutions in TBST. After that, the membranes were washed and incubated with secondary antibody for 1 h at room temperature. The amount of detected protein was assessed using ECL detection reagents (WBKLS0050; Millipore), and the immunoactivity band was detected by a fully automatic chemiluminescence image analysis system (Tanon-5200; Tanon Science & Technology, Shanghai, China). The levels of target proteins were normalized to GAPDH or β-actin, which served as a loading control. Each immunostaining band was visualized and quantitated by an analysis software named Image J (National Institutes of Health, Boston, MA, USA).
Oxygen Consumption Rate (OCR)
Oxygen consumption rates were measured by an XFe96 extracellular flux analyzer (Seahorse Biosciences, North Billerica, MA). In short, cells were seeded in a 96-well plate in DMEM/DF12 medium at a density of 3 × 105 cells/ cm2 before measurement. One hour before measurements, the cell culture medium was replaced with 180 μl FX assay medium (#102365-100, Seahorse Biosciences) composed of 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine at pH7.4, and then incubated at 37°C under normal atmosphere. XF cell Mito Stress Test Kit (#103015-100, Seahorse Biosciences) was used in the FX assay medium. Measurements were obtained at 37°C. This assay procedure included mixing (3 min), waiting period (2 min) and measurement (3 min). Oxygen consumption rate (OCR) was analyzed using wave software provided by Seahorse Biosciences. Basal respiration, maximal respiration, and proton leak were determined by XF Cell Mito Stress Test Generator that was provided by Seahorse Bioscience.
Real-Time RT-PCR
Total RNA of RVLM was extracted using extraction reagent and converted into first-strand cDNA (TaKaRa Biotechnology, Co., Ltd., Dalian, China) in accordance with manufacturer’s instructions. The mRNAs of CD86, CD206, σ-1R, IL-1β, TNF-α, IL-10, TGF-β, NLRP3, ASC, Caspase-1, β-actin and GAPDH were analyzed by quantitative real-time PCR. The sequences of primers were listed in Table S1. For normalization and relative quantification, the housekeeping gene β-actin/GAPDH was used as the internal standard reference gene. The relative quantification of gene expression was expressed as fold-change via normalization against β-actin by using the 2ΔΔCT method. Three tissues of three rats were used in each group. A 1-2 fold change versus normal controls was considered significant.
Table S1
The primer sequences used of the target genes measured
|
Gene
|
Primer
|
|
Forward
|
Reverse
|
|
IL-1β
TNF-α
TGF-β
IL-10
CD86
σ-1R
NLRP3
ASC
Caspase1
β-actin
GAPDH
|
5′-TGACCGAGATGTGGAGG-3′
5′-TTCTCATTCCTGCTCGTGG-3′
5′-ATGCCGCCCTCGGGGCTGCGGCTGC-3′
5′-CTGCTATGTTGCCTGCTCTT-3′
5′-CACCCGAAACCTAAGATGT-3′
5′-TGAGAGACCACGGGGACCTG-3′
5’-TTTCTGACCCTCGTAGTCTATCCA-3’
5’CCCCATAGACCTCACTGATAAA-3’
5’-CTTCTTTGTTCTGCGTCTGC-3’
5′-ACAGCTTCTTTGCAGCTCCTTCG-3′
5′-GTGCTGAGTATGTCGTGGAGTCTAC-3′
|
5′-GAATGGCAATGATGAGGT-3′
5′-CCGCTTGGTGGTTTGC-3′
5′-TACGGCGGGAGCCCCGACGCCGACG-3′
5′-GGTCAGTCGGTCTGGGTGTA-3′
5′-TGAAAGAGAGAGGCTGTTGGA-3′
5′-GCACCACACTGACTCTTCCATTC-3′
5’-GGCTGTGGACAATGGGAAGG-3’
5’-GCCCATAGCCTTCCCGCACT-3’
5’-ATTTGTCCCTTAGAAACAGC-3’
5′-ATCGTCATCCATGGCGAACTGGTG-3′
5′-GGCGGAGATGATGACCCTTTTGG-3′
|
Mitochondrial ROS measurement
Mitochondrial ROS were measured following the manufacturer’s instructions of MitoSOX Red kit (Thermo Fisher Scientific). Briefly, treated cells cultured on glass were washed and incubated with 0.5 μM MitoSOX. After reaction, cells were washed and observed by an inverted fluorescence microscope for detection of superoxide anion (red fluorescence). Samples without MitoSOX Red Reagent were used to subtract the background. Mean fluorescence intensity was determined, and all samples were normalized to control samples.
Endoplasmic Reticulum (ER) [Ca2+]ER and Mitochondrial [Ca2+]M Measurement
Cultured primary microglial cells were transfected with the mitochondria matrix targeted Ca2+ fluorescent sensor CEPIA4 and ER lumen-targeted Ca2+ fluorescent sensor GECO protein[36, 38, 39]. For mitochondrial Ca2+ measurements, primary microglial cells were transiently transfected with the plasmids pCMVR-CEPIA4mt targeted to the mitochondrial matrix 48 h before the experiments. To measure Ca2+ of ER, primary microglial cells were loaded with ER lumen-targeted Ca2+ fluorescent sensor GECO protein in normal culture media and/or prorenin-treated culture media for 2 or 3 days before imaging. For the capture of subcellular ER and mitochondrial Ca2+ signals, time-lapse or snapshot images of Ca2+ signals were obtained using confocal microscope (LSM800, Carl Zeiss Microscopy Ltd, Cambridge, MA) equipped with a Lambda DG4 illumination system (Sutter Instrument). Cells were illuminated at 440 nm (440AF21; Omega Optical) through a 455DRLP dichroic mirror, and emission was collected alternatively at 480 nm (480BP10; Omega Optical) and 535 nm (535AF26; Omega Optical), using a cooled, 12-bit CCD camera (CoolSnap HQ; Ropper Scientific). Cells were washed briefly at room temperature with intracellular-like medium (ICM) containing: 125 mM KCl, 19 mM NaCl, 10 mM Hepes (pH 7.3 with KOH), and 1 mM EGTA, 10mM glucose, pH-adjusted at 7.45 with NaOH.
Statistical analysis
Data were expressed as means ± SEM (standard error of mean). Statistical differences between the SIH rats treated with SKF10047, prorenin and/or SKF10047 treated microglia were analyzed by Student's t test. For comparison between multiple groups, one-way or two-way analysis of variance with repeated measurements was used to determine the differences between groups. When an ANOVA was significant, post hoc testing of differences between groups was performed using the least significant difference (LSD) test. P < 0.05 indicated that the differences were statistically significant. Statistical analysis was performed using GraphPad Prism (GraphPad Software, Version 6.0, La Jolla, CA, United States).
{kind=link}