2.1 A high-quality whole-genome assembly and annotation of R. cerealis Rc207
We generated 178,680 clean reads with mean length of 34.1 kb and N50 length of 44.8 kb by using Oxford Nanopore long-read sequencing. After a de novo assembly by using the filtered and polished subreads of the Nanopore sequencing, and a further polishing/correcting by using ~ 32 million Illumina NGS reads (from six libraries), we obtained the final high-quality R. cerealis Rc207 genome assembly consisted of 55 scaffolds with a total length of 56.36 Mb, a max scaffold length of 3.52 Mb, an N50 scaffold length of 1.68 Mb, and a GC content of 48.63% (Table 1). Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis revealed that the genome assembly contained 91.03% (264/290) complete BUSCOs, of which 90.53% (239/264) were single-copy (Table S1). Mapping with the Illumina short reads showed that 90.16% of the reads were properly mapped (Table S1).
Table 1
Summary of R. cerealis RC207 genome assembly and annotation
| Item | Result |
| Nanopore clean read | |
| - read number | 178,680 |
| - total length (Gp) | 6.09 |
| - mean length (bp) | 34,095 |
| - N50 length (bp) | 44,799 |
| Genome assembly | |
| - assembly size (Mb) | 56.36 |
| - scaffold number | 55 |
| - scaffold N50 (Mb) | 1.68 |
| - scaffold N90 (Mb) | 0.53 |
| - scaffold Max (Mb) | 3.52 |
| - GC content (%) | 48.63 |
| - Repeat sequence (%) | 17.87 |
| Protein-coding genes | |
| - gene number | 14,433 |
| - average gene length (kb) | 2.2 |
| - average CDS length (kb) | 1.54 |
| - average intron number | 6.32 |
| Non-coding RNAs | |
| - rRNA number | 60 |
| - rRNA family | 4 |
| - tRNA number | 138 |
| - tRNA family | 46 |
| - other ncRNA number | 42 |
| - other ncRNA family | 13 |
Approximately 17.87% of the sequences of the assembly were identified as repeat sequences, including 16.12% Class I (retro-transposons) and 1.49% Class II (DNA transposons) transposable elements (TEs, Table S2). A total of 14,433 protein-coding genes were predicted, with an average length of 2.20 kb, and an average intron number of 6.32 per gene (Table 1). A total of 10,222 (70.82%) of the predicted proteins could be mapped into an ortholog group using the OrthoMCL database, and 95.07% (13,721) of them could be annotated in different databases, including GO (15.31%), KEGG (26.87%), KOG (46.22%), Pfam (64.04%), Swissprot (51.02%), TrEMBL (94.75%), and NCBI-nr (94.91%). Hence, as much as ~ 5% of these proteins might be specific to the R. cerealis Rc207 (Table S3).
Further functional annotation showed that the R. cerealis Rc207 genome contained an arsenal of potentially diverse pathogenicity factors, such as: 1,080 secreted proteins, 822 carbohydrate-active enzymes (CAZymes), 461 proteases, 511 protein kinases, 38 mitogen-activated protein kinase (MAPK) pathway genes, 281 transporters including 89 ABC transporters, 227 transcription factors, 225 nucleases, 55 GTPases, 8 G-protein-coupled receptors (GPCRs), 89 lipases, 3,659 pathogen-host interaction (PHI) proteins, 197 cytochrome P450 (CYP450) proteins, and 196 fungal virulence factors (FVFs) (Table S4). Also, we predicted 165 genes that belong to 15 secondary metabolite-associated gene clusters, including one non-ribosomal peptide synthetases (NRPS) type cluster, six terpene synthases type clusters, and eight NRPS-like type clusters (Table S5). All of these proteins may play significant roles in pathogenesis of R. cerealis, and might also be associated with the exclusive necrotrophic lifestyle and the adaptation of this pathogen to an unique ecological niche.
2.2 SEM and transcriptomic analysis during R. cerealis Rc207 infection in wheat
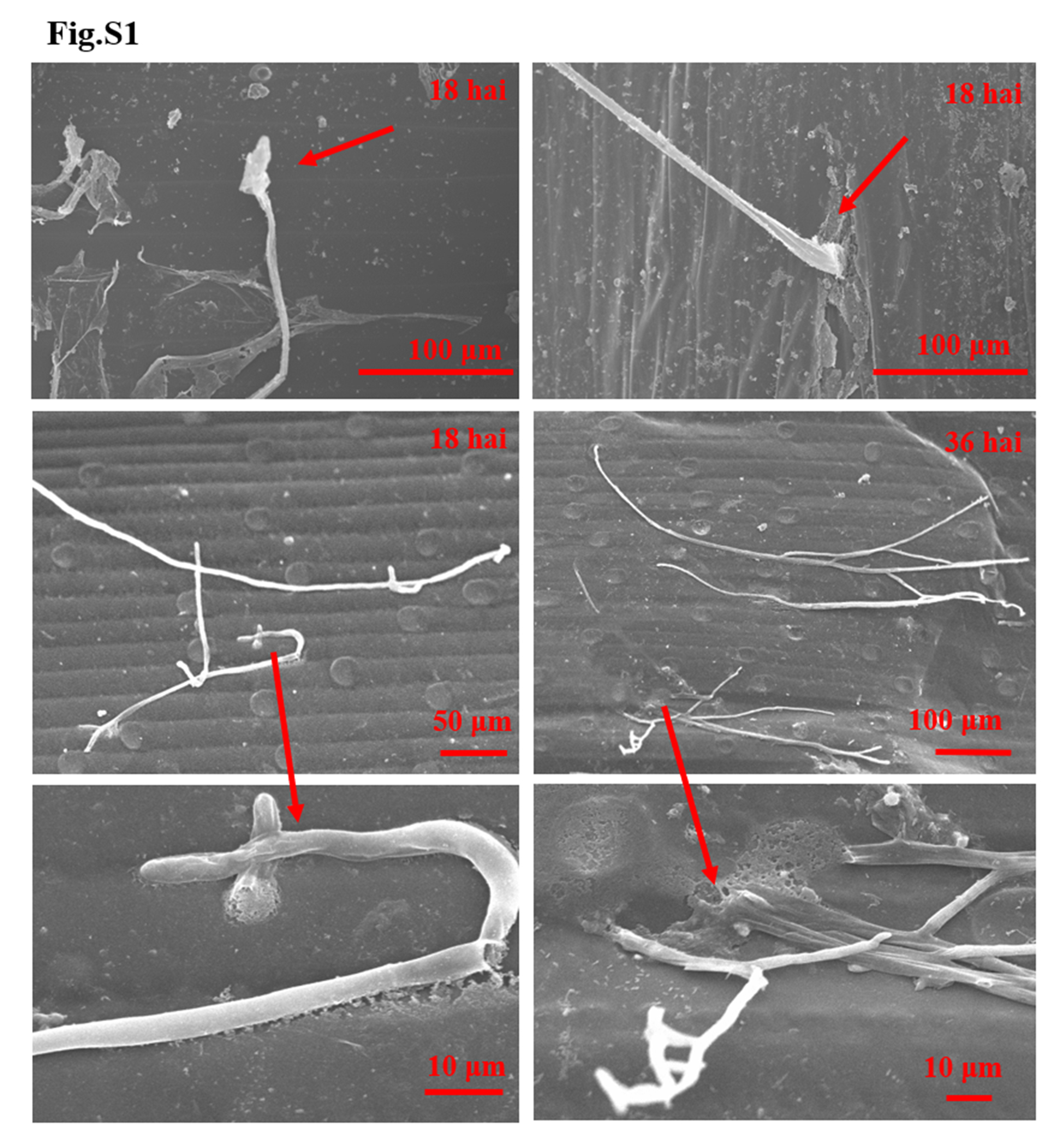
We used scanning electron microscopy to track the hyphae infection of R. cerealis Rc207 strain (on and) inside the leaf sheaths of the susceptible wheat cultivar Wenmai 6 at the tillering stage. At 18 hai, the hyphae started to pierce plant cell walls (Fig. 1A, Fig. S1). At 36 hai, the hyphae penetrated into the plant cell walls and spread into the cytoplasm of the infected wheat sheaths (Fig. 1A, Fig. S1). At 72 hai, the hyphae markedly proliferated on the surface of plant cells, and colonized and grew inside the invaded wheat cells (Fig. 1A). At the same time, small brown lesions were first visible at the surface of the infected leaf sheaths. At 96 hai, the thriving fungal hyphae occupied the whole plant cells and destroyed the cell walls (Fig. 1A), while the dark-brown lesions on the wheat leaf sheaths had expanded and continued to develop (Fig. S2). At 240 hai, the fungal hyphae massively proliferated inside the colonizing plant cells and continued to destroy the host cells, while the sharp eyespot symptoms on the inoculated wheat sheaths and stems became more severe (Fig. S2).
To investigate how the fungal transcriptional reprogramming occurs during wheat infection, we performed deep RNA-sequencing to investigate the global gene transcript dynamics of the R. cerealis Rc207 strain at five infection time-points (18, 36, 72, 96, and 240 hai) and in vitro mycelia. The transcriptomes showed that a total of 12,706 genes were expressed, and that 7,212, 6,931, 8,841, 12,071 and 12,085 genes were expressed at 18, 36, 72, 96, and 240 hai, respectively (Table S6). Compared with in vitro mycelia, a total of 70, 78, 495, 661 and 663 genes were significantly up-regulated (log2fold-change > 1, FDR P < 0.05) at 18, 36, 72, 96 and 240 hai, respectively (Table S7, Fig. 1B). The corresponding proteins of the 912 genes that were significantly up-regulated during infection belonged to a total of 529 OrthoMCL-annotated ortholog groups and 40 paralog groups, but 58 proteins could not be grouped. When compared to the whole genome, we found that 13 ortholog groups were significantly over-represented among the 912 genes (Table S8). Nine of these groups contained the CAZymes of Glycoside Hydrolase (GH) families 3 (OG6_100201), 6 (OG6_109747), 10 (OG6_102896), 32 (OG6_101843), 43 (OG6_110274), 51 (OG6_109774), and 61 (now is Auxiliary Activity family 9 or AA9; OG6_118077), carbohydrate-binding module (CBM) family 5 (OG6_100121), and AA7 (OG6_113017). Moreover, three of the groups contained aromatic peroxygenases (OG6_118111), malate dehydrogenase (OG6_123068), and sterol 14-demethylase (Cytochrome P450 family 51; OG6_103408). One of the groups contained stress response-related proteins (OG6_111600). Almost the entire set of protein groups belonged to the predicted secretome (Table S8), indicating that significantly up-regulated secreted proteins may play important roles during the interactions between R. cerealis and wheat, and that these secreted proteins may be major pathogenesis determinants .
2.3 Up-regulated secretory CAZymes contribute to the infection of R. cerealis
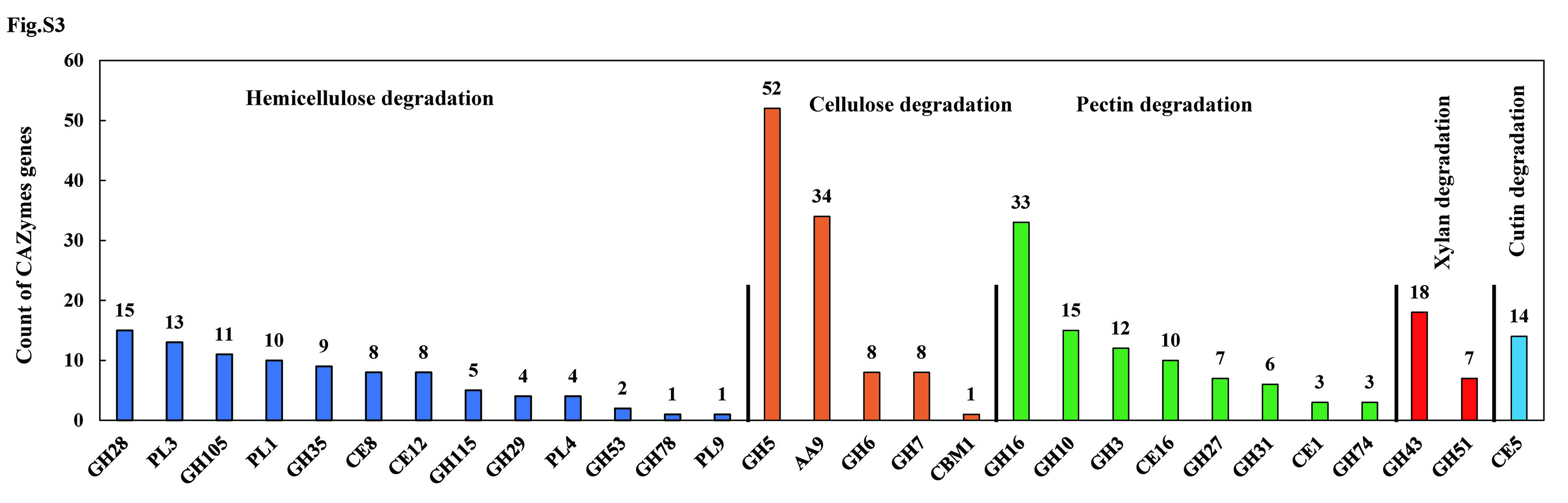
The R. cerealis Rc207 genome contained 822 genes encoding CAZymes that potentially degrade the structure of the plant cell wall and modify the fungal cell wall (Table S9). Among these, 404 CAZymes belonged to secretory proteins. Compared with 8 previously sequenced pathogenic fungi (Cryptococcus gattii, Fusarium graminearum, Melampsora larici-populina, Postia placenta, Puccinia graminis, R. solani AG1 IA, R. solani AG8, and Ustilago maydis), R. cerealis seems to be well equipped with more expanded gene families of CAZymes, and has the highest number in categories GH, carbohydrate esterase (CE) and AA (Table S10). Moreover, relatively to the rice sheath blight pathogen R. solani AG1 IA (399 CAZyme-genes), the R. cerealis RC207 genome contained a more abundant number of cellulose-degradation enzyme GH5 genes (52 vs. 15), hemicellulose-degradation enzyme GH16 genes (33 vs. 15), pectin-degradation enzyme GH28 genes (15 vs. 6), xylan-degradation enzyme GH43 genes (18 vs. 9), cutinase CE5 genes (14 vs. 1), chitin deacetylase CE4-coding genes (15 vs. 8), and AA9-coding genes (34 vs. 10) (Fig. S3, Table S11).
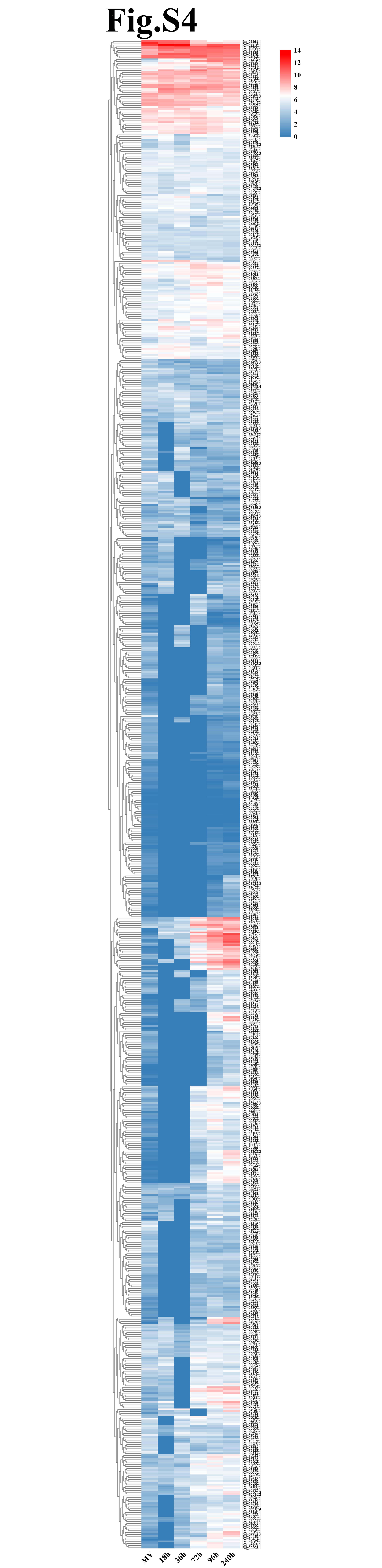
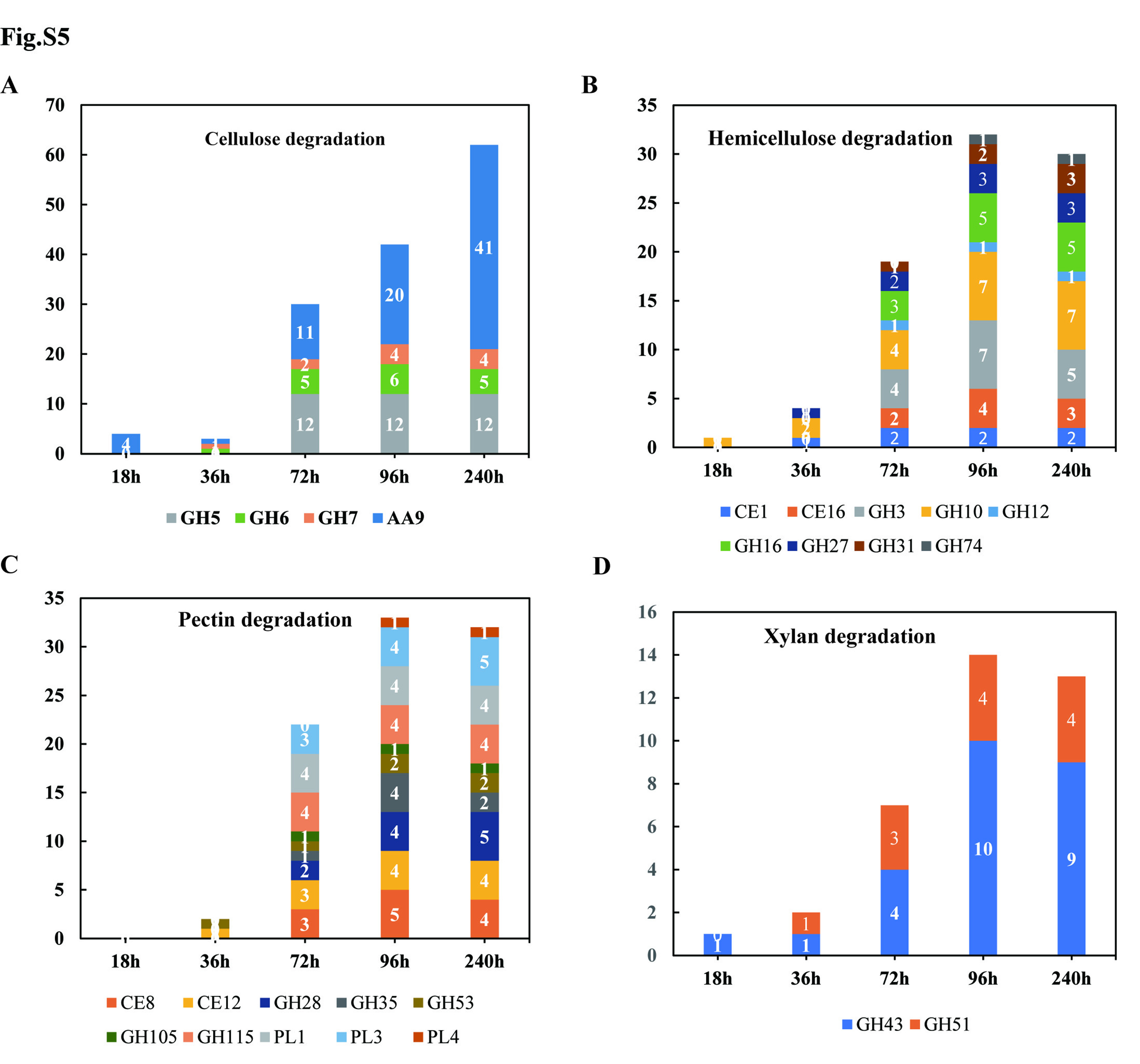
RNA-seq based expression profile analysis showed that, relative to the in vitro mycelia, 9, 15, 135, 208, and 202 CAZyme-coding genes were significantly up-regulated at 18, 36, 72, 96, and 240 hai, respectively (Fig. 2A, Table S12, Fig. S4). The transcript levels of 46 cellulose-targeted enzymes (GH5, GH7, and AA9), 34 hemicellulose-targeted enzymes (GH16, GH10 and GH3), 37 pectin degradation enzymes (GH28, PL1 and PL3), and 14 xylan-degradation enzymes (GH43 and GH51) were significantly up-regulated during the infection process (Fig. S5A-Fig. S5D). Additionally, 5 chitin deacetylase CE4-coding genes were significantly up-regulated during advanced infection stages (72 to 240 hai).
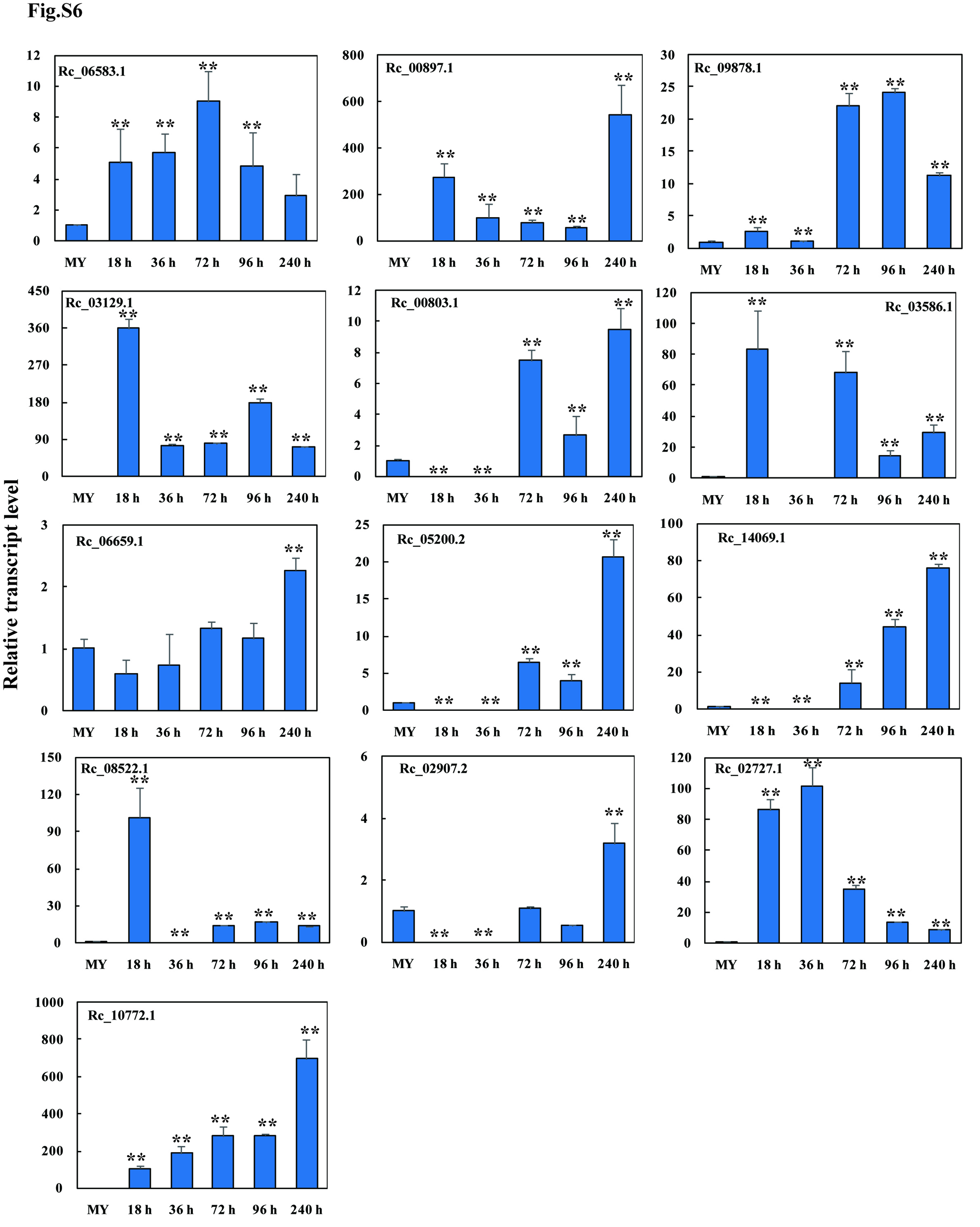
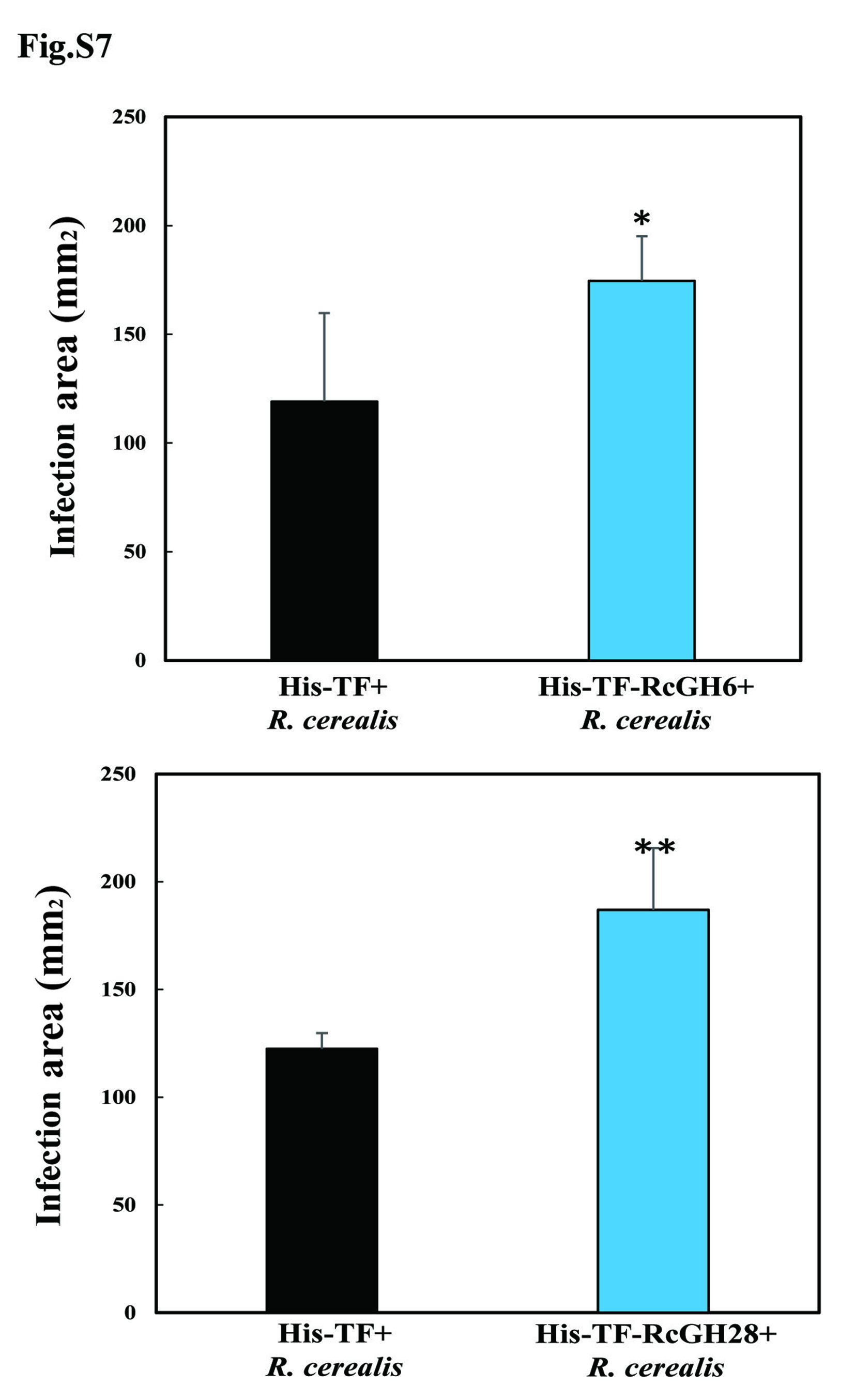
To further test an association between the up-regulated CAZyme-coding genes and pathogenesis, we examined the transcript profiles of 13 genes by qRT-PCR analyses. The results showed that the transcript levels of these 13 CAZymes-coding genes, including 1 GH28, 3 GH5, 1 GH6, 3 GH10, 1 GH51, 1 AA9, 2 CBM1, and 1 CE5, were significantly induced during infection in wheat (18, 36, 72, 96, and 240 hai) compared to the in vitro fungal mycelia (Fig. S6). Further, the functional validation results showed that the heterologously-expressed proteins of two up-regulated and cysteine-rich secretory CAZymes, including RcGH6-1 (Rc_00803.1) and RcGH28 (Rc_03586.1) (Fig. 2B), were able to trigger plant cell death/necrosis by infiltration into the leaves of the susceptible wheat cv. Wenmai 6 (Fig. 2C). Importantly, through assay the infection size on the filtrated wheat leaves plus R. cerealis liquid mycelia inoculation, results showed that the heterologously-expressed RcGH6-1 and RcGH28 proteins were further verified to promote the fungal infection (Fig. 2D, Fig. S7).
2.4 Secretory effectors in R. cerealis Rc207 contribute to the infection
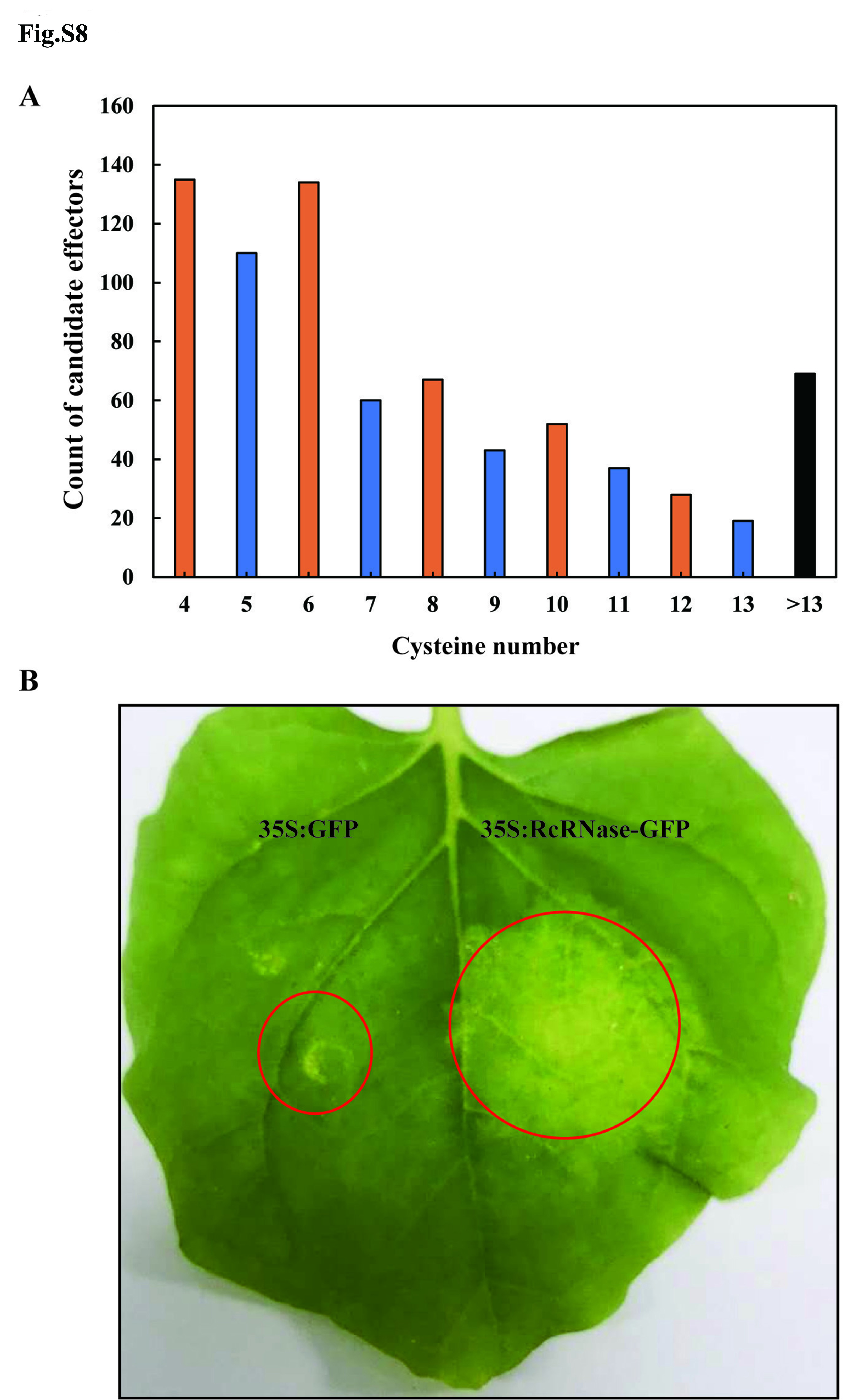
Although previous research annotated the candidate effectors with a signal peptide-containing protein pipeline17, experimentally-validated effectors and virulence factors tend to be cysteine-rich (≥ 4) or up-regulated secreted proteins15,19−21. The R. cerealis Rc207 secretome is comprised by 1,080 secreted proteins (Table S13). Transcriptome analysis showed that 300 genes coding for secreted proteins were significantly up-regulated during fungal infection in wheat (Table S14, Fig. 3A). Of these, we found that 177 and 105 genes coding even and odd cysteine-containing secreted proteins, respectively, were significantly up-regulated. Furthermore, the R. cerealis Rc207 secretome includes 755 cysteine-rich (the number of cysteine ≥ 4) secretory proteins, which likely function as candidate effectors (Fig. S8A). Taken together, a total of 831 candidate effectors, including 755 cysteine-rich secretory proteins and 282 up-regulated secretory proteins, were identified from the R. cerealis Rc207 ssecretome and classified according to their functional annotation (Table S14). Among them, those containing even cysteines outnumbered those containing odd cysteine numbers.
This set comprised 29 novel candidate effectors, including: alkaline phosphatases, antigens (carboxypeptidase Y inhibitor), cytochrome b2, choline transporter, glycerophosphoryl diester phosphodiesterase family, fruiting body protein SC7, tyrosinase, distantly related to plant expansin, allergen, alkaline nonlysosomal ceramidase, endoplasmic reticulum protein, FAD-dependent oxidoreductase, guanyl-specific ribonuclease, tripeptidyl peptidase, signal peptide-containing isochorismatase, and other 2 uncharacterized proteins that have obvious characteristics of effectors (Table S15). In addition, the R. cerealis Rc207 secretome also contained 1 cytochrome C oxidase (Rc_02038.1) with 13.21% amino acid identity to the oxidase domain of R. solani AG1 IA assembly protein CtaG/cox (AG1IA_05310, a novel effector reported)14, and three peptidase inhibitors I9 (Rc_08365.1, Rc_04605.1 and Rc_10139.1) with 28.57%, 25.94%, and 26.32% identities to the peptidase inhibitor domain of the R. solani AG1 IA peptidase inhibitor I9 (AG1IA_07795, a novel effector reported)14.
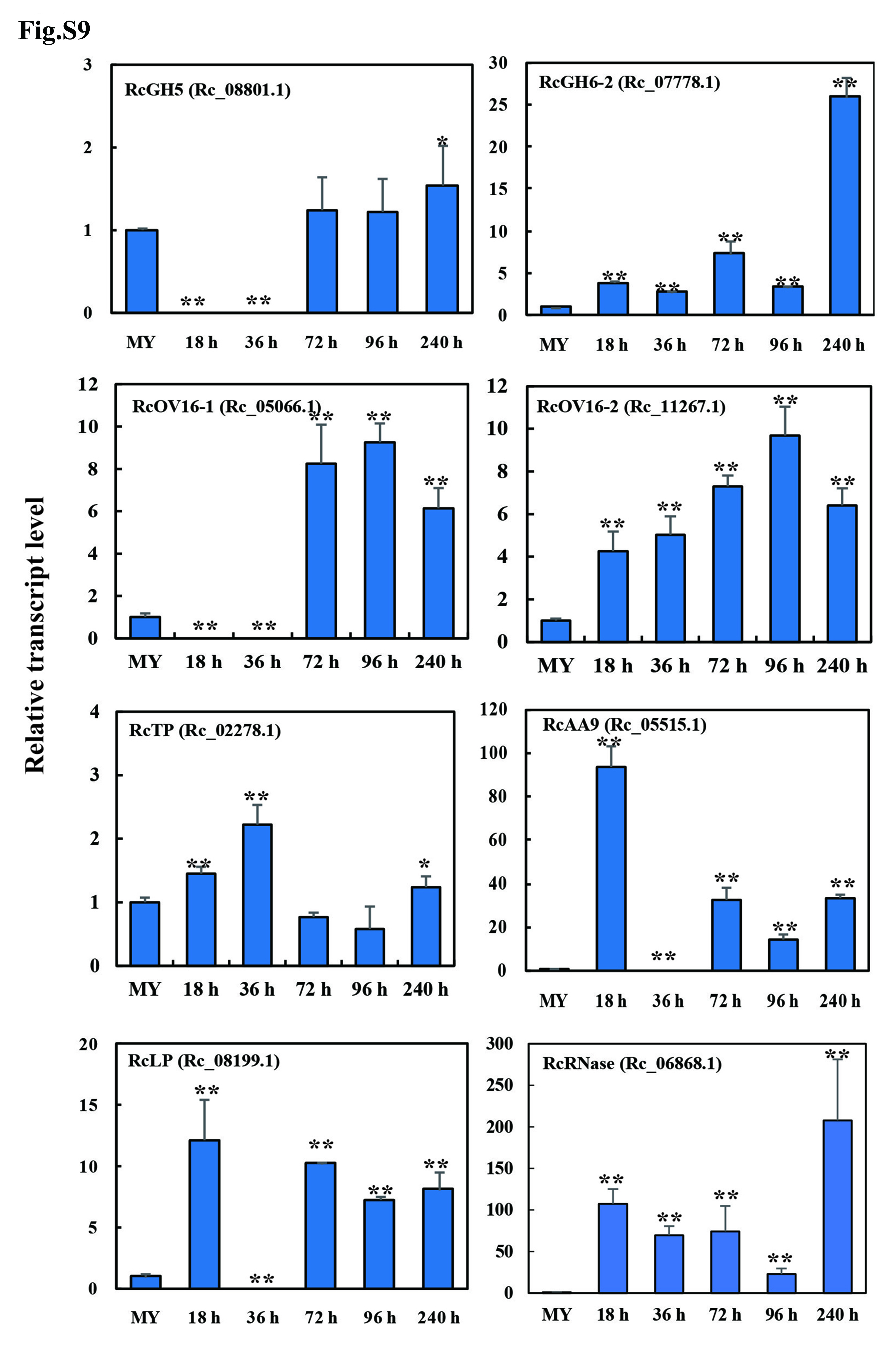
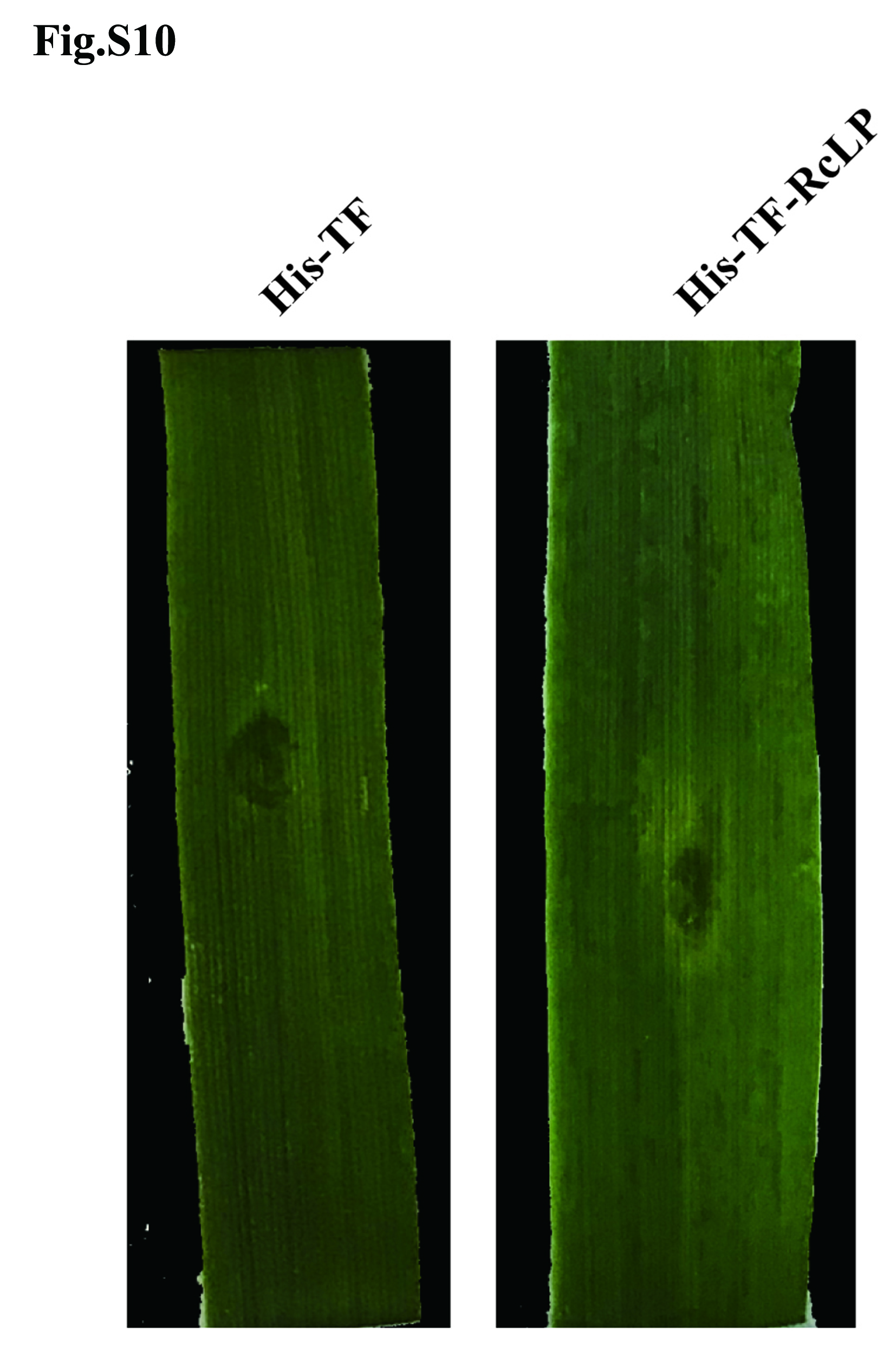
Previous studies showed that during necrotrophic fungal pathogen infection to host plants, secretory effectors function as essential determinants of pathogenicity or virulence through induction of plant cell death and necrosis22. We cloned and validated the functions of 11 up-regulated (qRT-PCR results, Fig. S6, Fig. S9, Fig. 4A), cysteine-rich candidate effectors with diverse activity during fungal infection in wheat, including: 5 CAZymes [RcGH5 (Rc_08801.1), RcGH6-1 (Rc_00803.1), RcGH6-2 (Rc_07778.1), RcGH28 (Rc_03586.1), and RcAA9 (Rc_05515.1)], 1 GDSL-lipase RcLP (Rc_08199.1), 1 metalloprotease RcFL1 (Rc_11192.1), 1 tripeptidyl peptidase RcTP (Rc_02278.1), 2 antigens RcOV16-1 and RcOV16-2 (Rc_05066.1 and Rc_11267.1), and 1 guanyl-specific ribonuclease RcRNase (Rc_06868.1). The results showed that except that the heterologously-expressed Rclipase (Rc_08199.1) did not obviously induce necrosis in infiltrated wheat leaves (Fig. S10), the remaining 10 secretory proteins were able to induce cell death in the treated leaves of wheat or N. benthamiana, and thus promoted R. cerealis Rc207 infection in host plants (Fig. 2C-2D, Fig. 3B-3D, Fig. 4C-4E).
2.5 Secretory metalloprotease RcFL1 is required for fungal infection in wheat
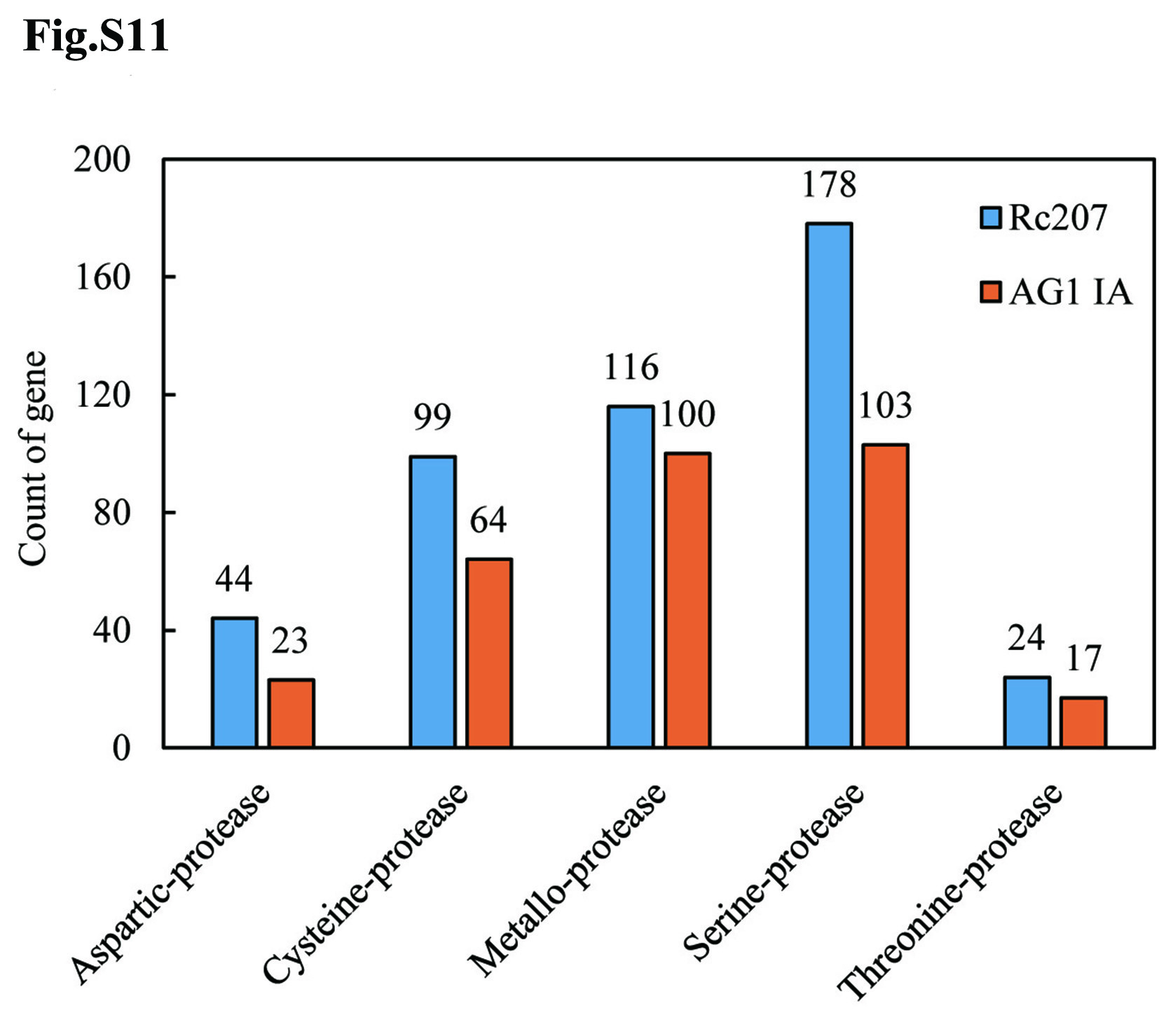
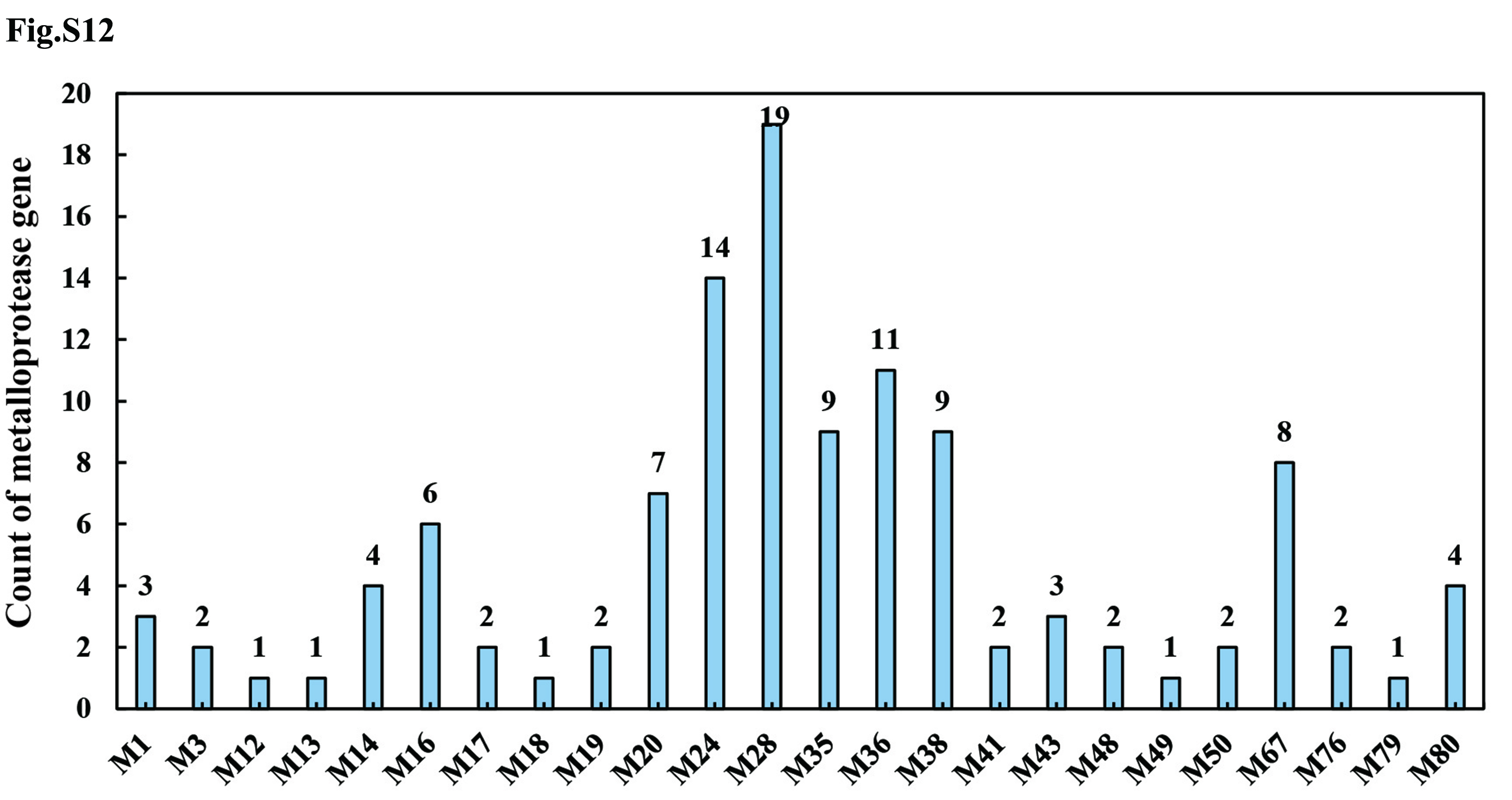
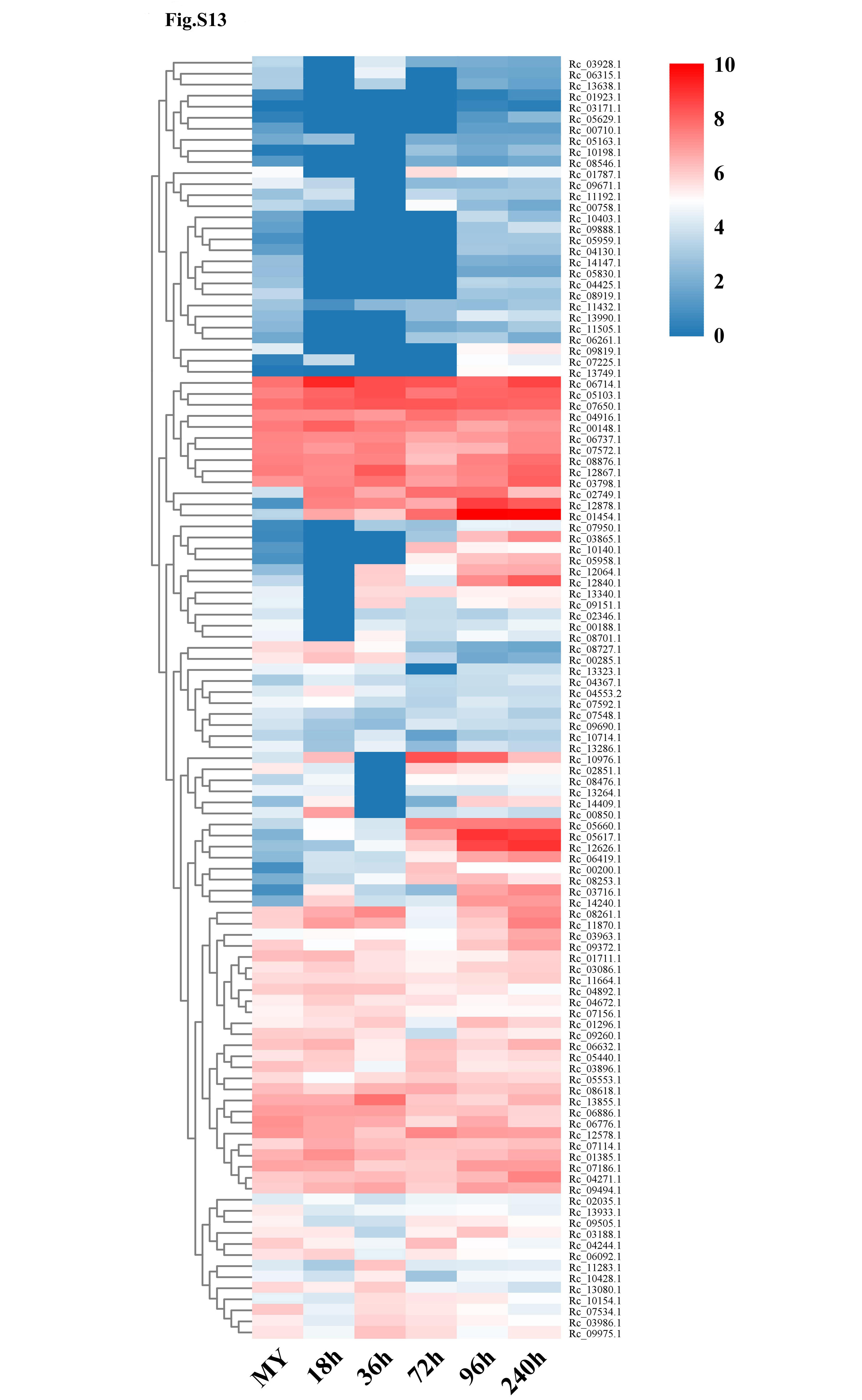
Compared to the closely-related fungus R. solani AG1 IA (307 protease-coding genes), the R. cerealis Rc207 genome contained more genes coding for proteases (461 genes), particularly richer in aspartic proteases, including pepsin A1As (44 vs. 23), cysteine proteases (99 vs. 64), metalloproteases (116 vs. 100), serine proteases such as prolyl aminopeptidase S33s (178 vs. 103), and threonine proteases (24 vs. 17) (Fig. S11, Table S16). Among the 116 metalloproteases (Fig. S12), 6, 4, 13, 19 and 19 metalloprotease-coding genes were significantly up-regulated during wheat infection at 18, 36, 72, 96 and 240 hai, respectively (Fig. S13, Table S17).
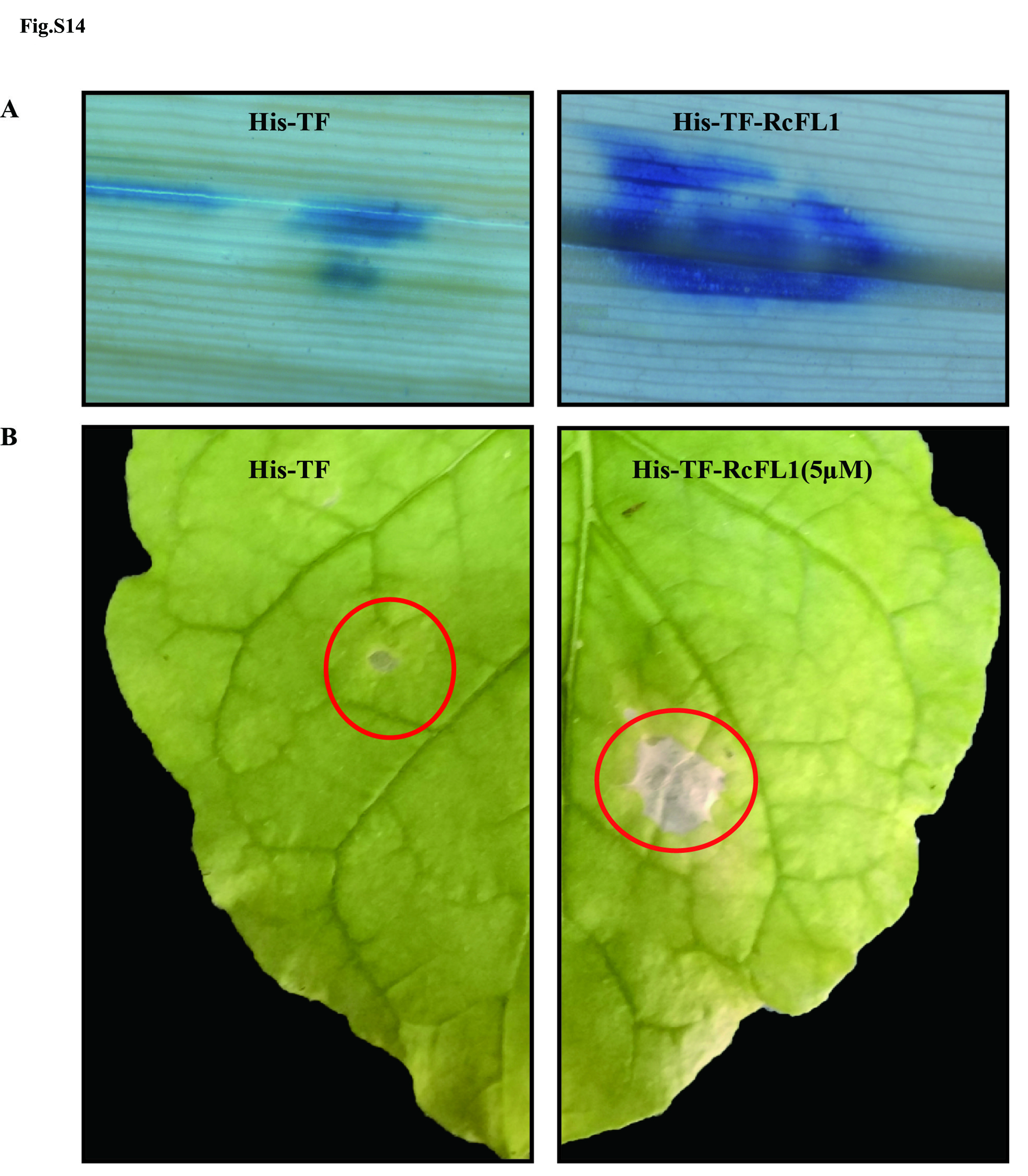
In order to further investigate an association between the up-regulated metalloproteases with pathogenesis, we examined the gene transcript profiles and the pathogenic functions of the M36 domain-containing metalloprotease (fungalysin) RcFL1 (Rc_11192.1) and M43 domain-containing metalloprotease RcMEP123, both with signal peptides, during R. cerealis infection in wheat. The qRT-PCR analysis results showed that the transcript abundances of RcFL1 and RcMEP123 were markedly up-regulated during the infection process in wheat (18, 36, 72, 96, and 240 hai) compared to in vitro fungal mycelia (Fig. 4A). Our previous functional analyses showed that the up-regulated secretory RcMEP1 acted as virulence factor23. Hence, we further investigated functional role of the RcFL1 during fungal infection to wheat. Agrobacterium tumefaciens mediated transient expression assays in Nicotiana benthamiana leaves showed that the RcFL1 was able to secrete into the apoplasts and to trigger plant cell death, but that the signal peptide-deleting mutant lost both of these activities in planta (Fig. 4B). Furthermore, compared to His-TF (CK), the heterologously-expressed proteins His-TF-RcFL1 (expressing the full RcFL1 protein) and His-TF-RcFL1-M36 (expressing the M36 domain of RcFL1) were able to trigger necrosis and plant cell death on the infiltrated leaves of susceptible wheat cv. Wenmai 6 or of N. benthamiana (Fig. 4C, Fig. S14A, Fig. S14B). Notably, both the RcFL1 and RcFL1-M36 proteins were able to increase the fungal infection size after R. cerealis liquid mycelia inoculation on the wheat leaves (Fig. 4D-4E). Additionally, the RcFL1 protein significantly repressed the expression of wheat defense genes encoding chitinases (TaChit3 and TaChitIV) and the receptor-like kinase CERK1 (TaCERK1) in the infiltrated leaves (Fig. 4F). These results demonstrated that the secretory metalloprotease RcFL1, acting as a virulence factor, is required for the fungal infection into wheat, and that both the signal peptide and the M36 domain are necessary to ensure the virulence role of RcFL1.
2.6 Repeat sequences and frequently-intraspecies gene duplication contribute to the genome evolution of R. cerealis and expansion of pathogenicity-related gene families
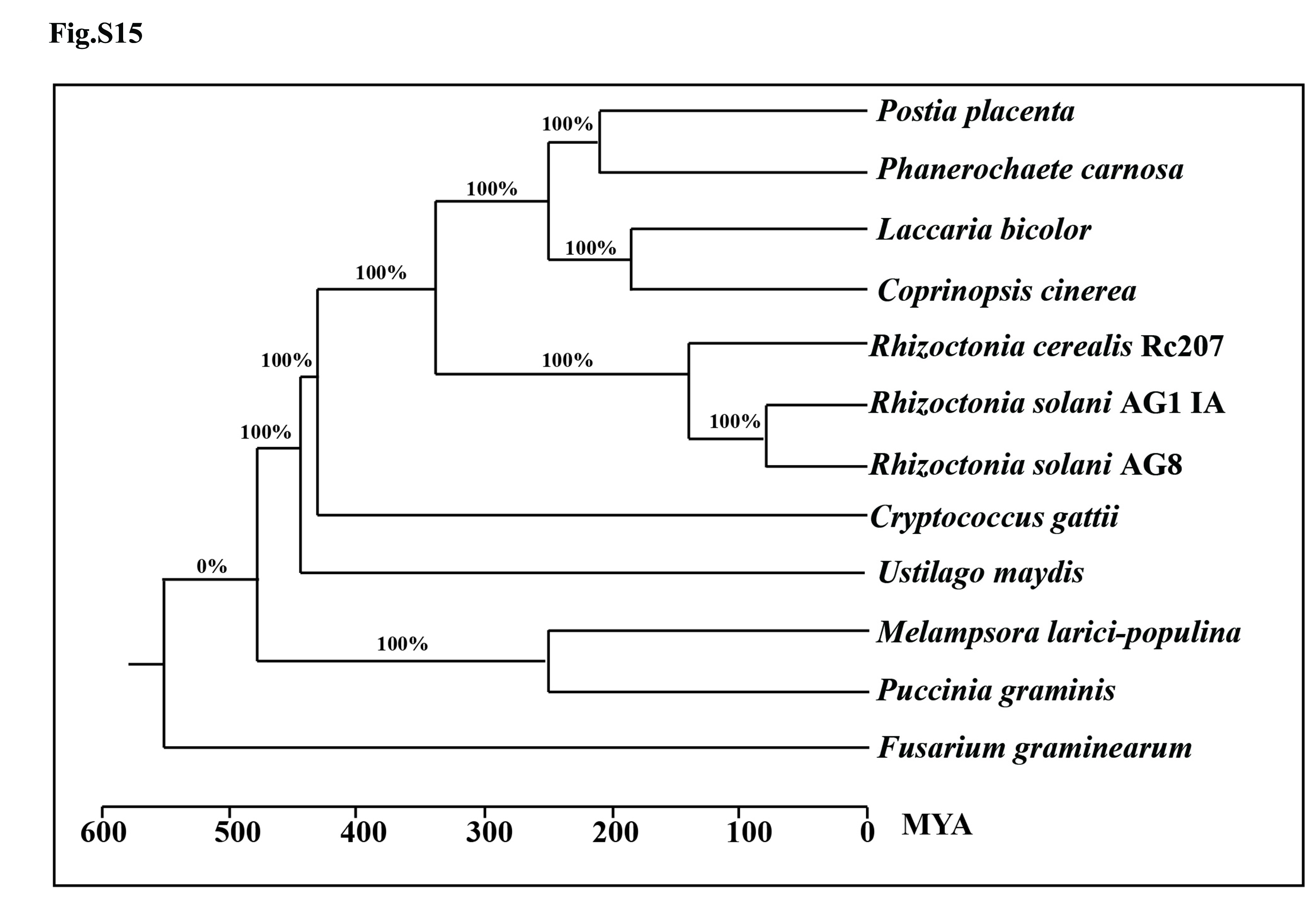
A phylogenetic tree including R. cerealis Rc207, ten previously sequenced basidiomycotina fungi (Coprinopsis cinerea, Cryptococcus gattii, Laccaria bicolor, M. larici-populina, Phanerochaete carnosa, Postia placenta, Puccinia graminis, R. solani AG1 IA, R. solani AG8, and U. maydis), and the ascomycete F. graminearum as an outgroup, was constructed based on their core orthologs. The tree showed that R. cerealis Rc207 is more closely related to R. solani AG1 IA (and R. solani AG8) than to the other fungi in the basidiomycotina family. The divergence between R. cerealis Rc207 and R. solani AG1 IA/R. solani AG8 occurred approximately 140 million years ago (MYA; Fig. S15).
To explore the major mechanisms underlying the genomic evolution of R. cerealis, we completed the genome assembled of R. cerealis Rc301 (R0301, another strain is also virulent to wheat isolated from Nanjing, China) by using Illumina reads (Table S18), then performed intra-species gene collinearity analysis between R. cerealis Rc207 and R. cerealis Rc301, and inter-species gene collinearity analyses between R. cerealis and R. solani AG1 IA/R. solani AG8. These analyses showed there were more collinear gene clusters identified from the longer scaffolds of R. cerealis Rc207 genome assembly (nanopore sequencing), indicating that these scaffolds include conserved regions. In contrast, more R. cerealis-specific the genes were present in the short scaffolds (Fig. 5A-5B). In addition, we identified 3,010 intra-species syntenic (IS) genes (20.86%) in 53 syntenic gene clusters within the R. cerealis Rc207 genome (Table S19). These IS genes exhibited a high frequency among the short scaffolds and a low frequency among the long scaffolds (Fig. 6), and the similar pattern was also observed in R. cerealis Rc301 genome (Fig. 5B). Interestingly, the distribution pattern of IS genes was similar to that of repeat sequences that usually accompany the genomic structures for a rapid evolution16,24,25. These findings suggest that there may be abundant duplicated genes and gene clusters within the R. cerealis genome, and that these genes are frequently located at repeat sequences-rich genomic regions that may be undergoing rapid evolution.
According to the lengths of the scaffolds, from long to short, and to the frequency of IS genes, with 4.6%, 21.6% and 40.3%, we divided the scaffolds into three groups to perform preliminary comparisons. Specifically, group1, group2 and group3 included the top five longer scaffolds, the 13 medium-length scaffolds, and the remaining 37 short scaffolds, respectively (Fig. 5B; Fig. 6, Table S21). In a manner consistent with the observations above on the repeat sequences, the IC genes, the secretome- and CAZymes-encoding genes and the species-specific genes, the unexpressed and low-expressed genes also exhibited an increasing frequency trend from group1 to group3. In contrast, an opposite trend was observed for gene density, GC content, the frequency of conserved genes and that of genes with collinearity to Rc301 (Fig. 5B, Fig. 6; Table S20). In addition, the IC genes, the species-specific genes, the unexpressed and low-expressed genes, and the repeat sequences, concentrated on the edge of scaffolds; while the conserved genes, the highly expressed genes, the majority of the secretome, and the genes with collinearity to RC301 concentrated on the central regions of scaffolds (Fig. 5B, Fig. 6A-C; Table S21). These analyses further revealed that repeat sequences likely drive a rapid evolution in specific regions of the R. cerealis genome, which may be in the forms of mobile chromosomes or plastic regions, such as the edge of the chromosomes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}