Melanoma patient serum samples
Study design, baseline clinical and demographic characteristics, assessments and definition of endpoints of a cohort of advanced stage IV melanoma patients was recently described in (1). Briefly, clinical data and sera of advanced stage IV melanoma patients that either responded (referred to as responders (R)) = partial response or complete response, n = 19) or failed to respond (referred to as non-responders (NR) = stable disease or progressive disease, n = 23) to combinatorial IFNa and aPD1 were used in this study (ClinialTrials.gov identifier: NCT02112032; KEYNOTE-020). Collected serum samples at baseline in treatment naïve patients were used to analyze systemic I3A and kynurenine by mass spectroscopy. Approval to treat patients was obtained from the University of Pittsburgh’s Hillman Cancer Center (HCC) Institutional Review Board (No. PRO14030075).
Animals
C57BL/6 mice were obtained from The Jackson Laboratory. IfngL E8I Cre mice were provided by Dr. Dario A. A. Vignali, University of Pittsburgh, and Rag2-/- mice were kindly provided by Dr. M. Shlomchik, University of Pittsburgh. Ahrfl CD8 Cre mice were generated by crossing Ahrfl (Ahrtm3.1Bra/J, The Jackson Laboratory, 006203) mice with CD8a-Cre (C57BL/6-Tg(Cd8a-cre)1Itan/J, The Jackson Laboratory, 008766) mice. For all experiments, 6-10 week-old females or males were used; no notable sex-dependent differences were found for the reported experiments. Mice were housed at the University of Pittsburgh animal facilities under specific pathogen-free (SPF) conditions, where cages were changed on a weekly basis. Ventilated cages, bedding, food and water (non-acidified) were autoclaved before use, ambient temperature maintained at 23 °C, and 5% Clidox-S was used as a disinfectant. Experimental and breeding cages were randomly housed on two different racks in the vivarium, and all cages were kept on automatic 12-h light/dark cycles. Animal care and experimentation were conducted in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Gnotobiotic animal husbandry
Food, bedding, and water (non-acidified) were autoclaved before transfer into the sterile isolators. Cages within isolators were changed weekly, and all the cages in the vivarium were kept on 12-h light/dark cycles. Microbiology testing of fecal (experimental mice) or of cecum samples (sentinel mice; aerobic and anaerobic culture, 16S qPCR) was performed every other week to confirm germ-free status.
Tumor models
On day zero mice were injected subcutaneously in the right hind flank with 106 B16-F0 (ATCC, CRL-6322), 106 YUMM1.7 (ATCC, CRL-3362), or 5 x 105 MC38 (Kerafast, ENH204-FP) tumor cells in 100 µL sterile PBS. Tumor volumes were calculated using the formula tumor volume= length x width2x 0.5, where length represents the largest tumor diameter and width represents the diameter perpendicular to the length (2). For survival experiments mice were sacrificed when tumors reached a volume of ³ 6000 mm3.
Administration of bacteria, I3A, KYN, and antibiotics
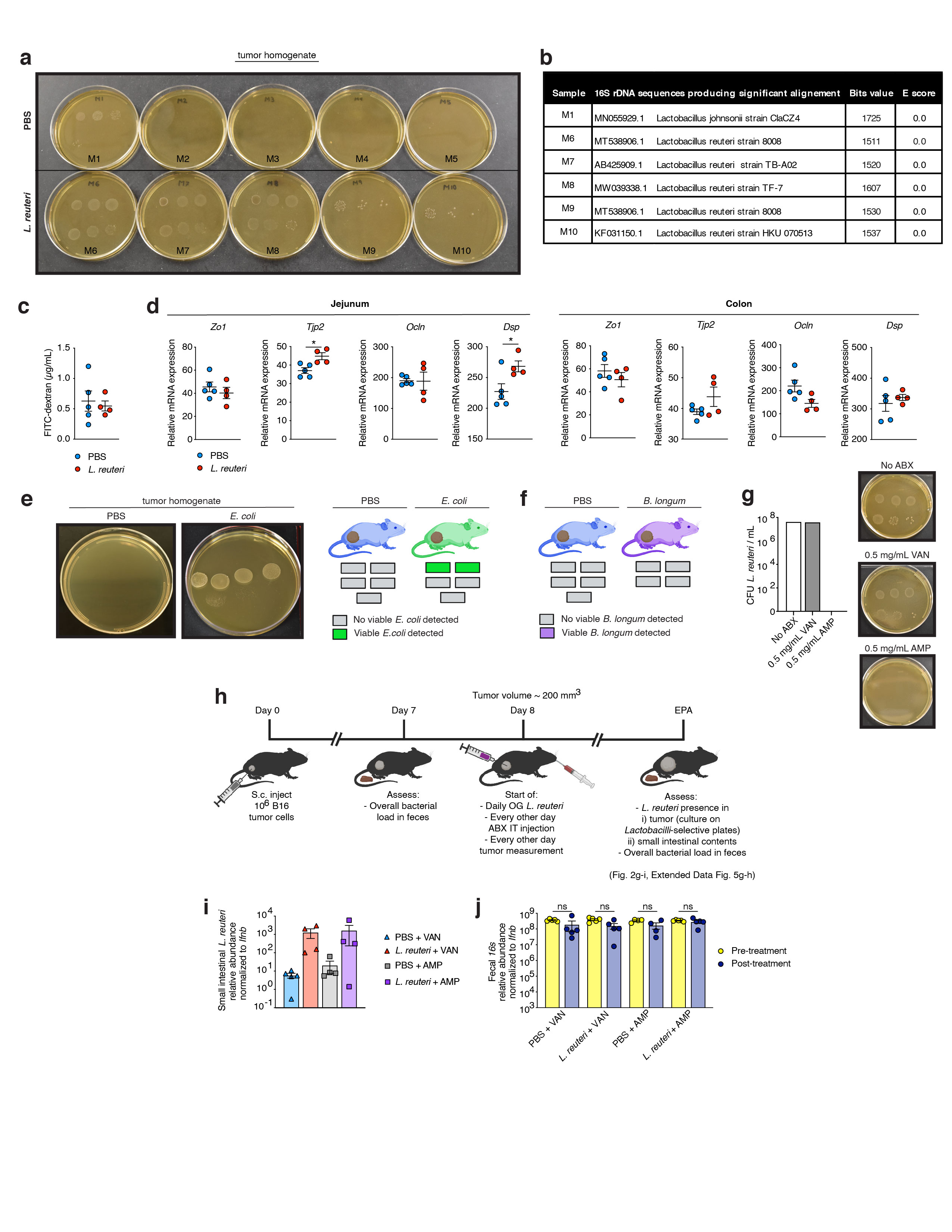
Lactobacillus reuteri DArAT lacking the gene coding for aromatic amino acid aminotransferase class I/II (3) was generously provided by Dr. L. Cervantes-Barragan, Emory University. Lactobacillus reuteri (ATCC, BAA-2837, isolated from human breast milk), Lactobacillus johnsonii (ATCC BAA-3147), Bifidobacterium longum Reuter (ATCC, BAA-999), and Lactobacillus reuteri DArAT were cultured anaerobically in MRS Broth (BD Difco, DF0881-17-5) at 37°C. Escherichia coli (ATCC, BAA-1429) was cultured aerobically in Tryptic Soy Broth (BD Bacto, DF0370-17-3) at 37°C. Briefly, for oral gavage experiments mice were gavaged daily with 109 colony forming units (CFU) bacteria in 200 µL PBS, or 20 mg/kg body weight (b.w.) or 40 mg/kg b.w Indole-3-Aldehyde (Sigma-Aldrich, 129445) in 200 µL corn oil, or vehicle control starting one day post tumor cell engraftment until endpoint analysis (EPA) unless noted otherwise. For heat-killed experiments, bacteria were incubated at 95 °C for 150 minutes (min) prior to gavage. For intratumoral injection experiments, mice were injected with 2x107 CFU viable or heat-killed L. reuteri or E. coli in 40 µL PTT, 10 µM, 1000 µM, or 200µg/mL I3A or L-kynurenine (Sigma-Aldrich, K8625) in 40 µL 10% Tween 20, 0.5 mg/mL ampicillin (Fisher BioReagents, BP1760-25) or vancomycin (Sigma-Aldrich, V2002) in 40 µL sterile water, or vehicle control starting when tumors reached an average volume of 300 mm3 every three days until EPA unless indicated otherwise.
In vitro assessment of ampicillin and vancomycin effectiveness against L. reuteri
5 x 107 CFU L. reuteri were inoculated in MRS broth containing 0.5 mg/mL ampicillin or vancomycin or vehicle control (sterile water). Cultures were grown anaerobically at 37 °C for 24 hours (h). Cultures were plated on MRS agar and incubated at 37 °C for 24 h under anaerobic conditions. CFU were then quantified.
CD8/CD4 T cell depletion and anti-PD-L1 mAb immunotherapy
For depletion of CD8/CD4 T Cells mice were injected intraperitoneally weekly with 250 µg InVivoMAb anti-mouse CD8a (BioXCell, BE0061), InVivoMAb anti-mouse CD4 (BioXcell, BE0003-1), or isotype control (BioXCell, BE0090) for a total of three times. For anti-PD-L1 mAb immunotherapy experiments, mice were injected intraperitoneally on day 5, 7, 9, and 12, with 100 µg InVivoMAb anti-mouse PD-L1 (BioXCell, BE0101) or InVivoMAb rat IgG2b isotype control (BioXCell, BE0090).
Tissue harvest and cell purification
Tumors and spleens were harvested with autoclaved tools under sterile conditions and weight was recorded. Spleens were mashed and underwent erythrocyte lysis using the Mouse Erythrocyte Lysing Kit (R&D Systems, WL2000) and remaining cells were used for flow cytometry analysis. Tumor-intrinsic lymphocytes were isolated via purification of mononuclear cells using 40% percoll centrifugation (Cytiva, 17089101), subsequent erythrocyte lysis, and an enrichment of CD45+ cells as described: cells were incubated for 5 min on ice with rat serum and Fc block (BD Biosciences, 553142), followed by a 15 min incubation on ice with biotinylated anti-CD45 (Biolegend, 103104). Cells were washed, and incubated with streptavidin beads (BD Biosciences, 557812) for 20 min, followed by a 5 min incubation in an EasySep magnet (STEMCELL, 18000). Cells poured out from the magnet were discarded, and cells remaining were used for flow cytometry analysis.
Flow cytometry
Single-cell suspensions were prepared as described above and resuspended in FACS buffer (PBS, 2% FCS) for immunostaining and subsequent FACS analysis. Cell suspensions were incubated with Fc Block (BD Biosciences, 553142), followed with surface marker antibody (Ab) stain for 20 min at 4 °C. Abs were used as follows: anti-CD45 (BV480, BD Biosciences, 566095), anti-TCRb (Alexa Fluor® 700, BD Biosciences, 560705), anti-CD4 (BV650, BD Biosciences, 563232), anti-CD8 (BV570, BioLegend, 100740), anti- I-A/I-E (BUV496, BD Biosciences, 750281), anti-Ly-6G/Ly-6C (Alexa Fluor® 700, BioLegend, 108422), anti-CD11c (FITC, BioLegend, 117306), anti-CD11b (APC-eFluor 780, eBioscience, 47-0112-80), anti-F4/80 (PE-Cyanine5, eBioscience, 15-4801-80). For dead cell exclusion, cells were stained with Zombie NIR Fixable Viability dye (BioLegend, 423105) for 10 min at 4 °C and washed in FACS buffer. For intracellular cytokine and transcription factor staining, surface Ab-stained cells were first fixed and permeabilized using the FoxP3 Transcription Factor Staining Buffer kit (eBioscience, 00-5523-00) following manufacturer’s instructions. Cells were further stained with Abs against intracellular proteins for 30 min at 4 °C. Abs were used as follows: anti-IFNg (BV605, BioLegend, 505839), anti-FoxP3 (FITC, eBioscience, 11-5773-82), anti-Granzyme B (PE, eBioscience, 12-8898-80), anti-Ki67 (PE-eFluor 610, eBioscience, 61-5698-82). Samples were FSC-A/SSC-A gated to exclude debris and gated to exclude dead cells. Samples were run on an Aurora (Cytek) flow cytometer and analyzed with FlowJo 10 (Tree Star).
Single-cell RNA sequencing sample preparation
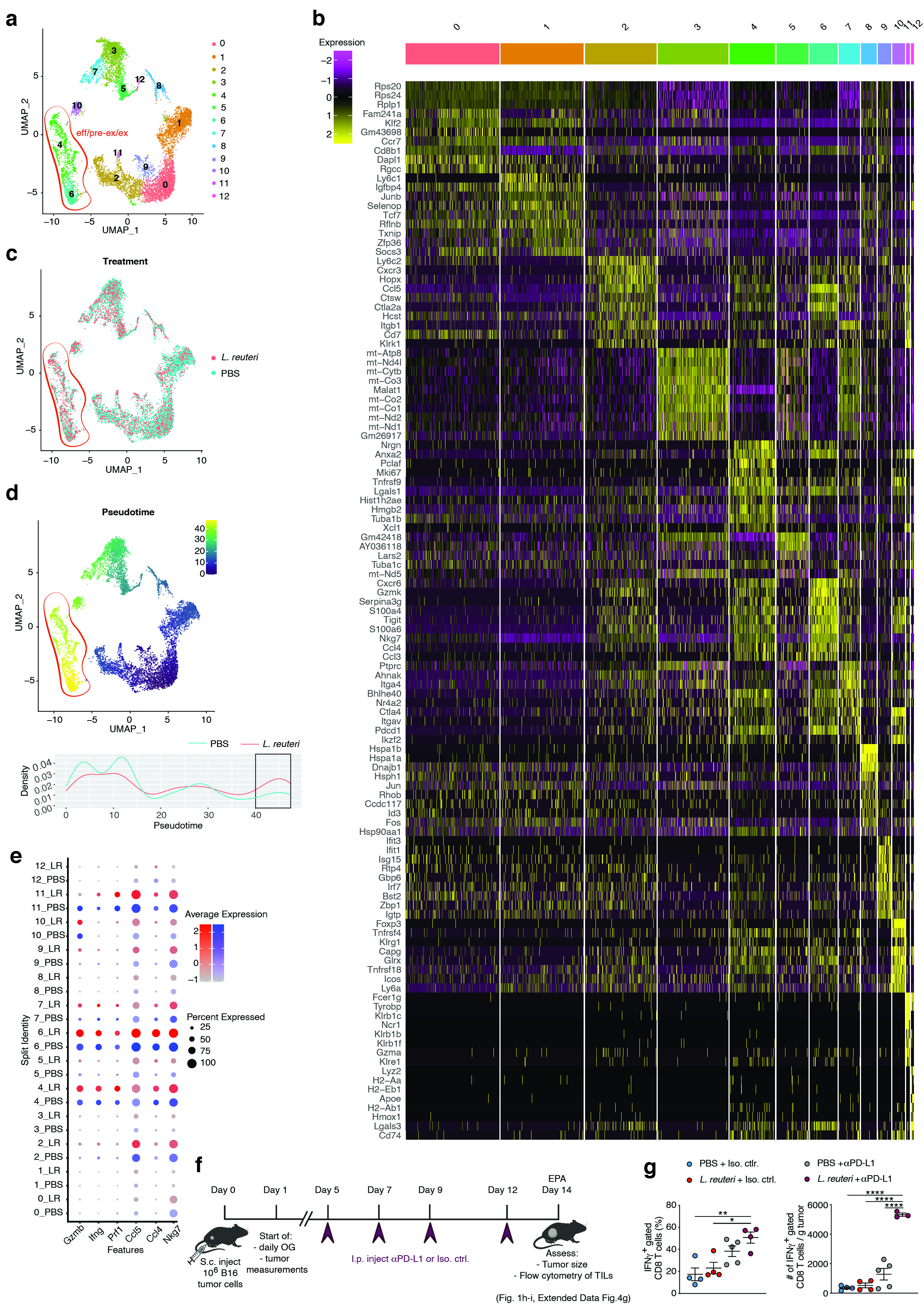
Single-cell suspensions from tumors (n = 4 per condition) were prepared as described above except as follows. Cells were purified via an enrichment for CD90.2+ cells using EasySep™ Mouse CD90.2 Positive Selection Kit II (STEMCELL, 18951). Samples were stained using anti-TCRβ (Alexa Fluor® 700, BD Biosciences, 560705), anti-CD4 (BUV395, BD Biosciences, 563790), anti-CD8a (BUV737, BD Biosciences, 612759), and each sample was stained with TotalSeq™-C03NN anti-mouse Hashtag 1 Antibodies with unique hashtag oligonucleotides (HTOs) in staining buffer for 30 min on ice (Biolegend Cat # 155861, 155863, 155865, 155867, 155871, 155873, 155875, 155877). For dead cell exclusion, cells were stained with LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Invitrogen, L34957) for 10 min on ice. Doublets and dead cells were excluded. From each animal, 8,000 CD8+ live cells were sorted using a 5-laser Aria-10 cytometer, and samples from the same condition were pooled in PBS + 0.04% BSA. GEM formation and library preparation was performed using Chromium Next GEM Single Cell 5' v2 workflow from 10x Genomics (Chromium Next GEM Single Cell 5' Kit v2, 16 rxns PN-1000263, Library Construction Kit, 16 rxns PN-1000190, 5' Feature Barcode Kit, 16 rxns PN-1000256, Chromium Next GEM Chip K Single Cell Kit, 48 rxns PN-1000286, Dual Index Kit TT Set A, 96 rxns PN-1000215, Dual Index Kit TN Set A, 96 rxns PN-1000250). Hashtagged cells from 8 mice (4 PBS treated and 4 L. reuteri treated) were pooled onto a single lane of the 10x chip to allow sample multiplexing. Prepared libraries were quality-checked on Agilent TapeStation and sequenced on HiSeq4000 (Novogene Inc).
Single-cell RNA sequencing analysis
Sequencing data were downloaded from Novogene onto the Joglekar laboratory server. Reads were aligned with CellRanger-4.0.0 to the Mus musculus reference genome (mm10-2020-A). Hashtag oligonucleotide sequences were added in the feature reference file included in the cellranger count step. Following alignment and generation of gene expression matrix, samples were processed in Seurat v4.0.1 (4). We used Seurat’s hashtag demultiplexing workflow to read hashtag oligos (HTODemux). Data from the resultant HTOs were re-processed and re-demultiplexed for further analysis. First, data were normalized, and integration anchors were identified. Integrated data were scaled and used for downstream Principle Component Analysis and visualization using Uniform Manifold Approximation Embedding (UMAP). First 20 principle components were used to derive Seurat clusters. Differential gene expression analysis was performed using the FindMarkers function in Seurat using negbinom testing. Trajectory analyses were performed using Monocle 3 (5, 6). Visualization and all statistical testing was performed in RStudio running on R version 4.0.3.
In vivo intestinal permeability measurement
To determine intestinal epithelial barrier permeability in mice, FITC-labelled dextran method was used as previously described (7). In brief, mice were withheld from food and water for 4 hours upon which they were gavaged with 60mg/100g body weight Fluorescein isothiocyanate (FITC)–dextran (MW 4,000, Sigma-Aldrich, 46944). After 3 h blood was collected by cheek bleeding, spun at 10,000 rpm for 10 min at 4 C, and 50 µL plasma was added to a 96-well flat bottom plate (Corning, 07-200-656) in duplicate. Analysis of FITC–dextran concentration was performed with a fluorescence spectrophotometer setup (SpectraMax® i3x using SoftMax Pro 3.0.7 Software) under the following settings: read type endpoint at excitation wavelength 485 and emission wavelength 520 (bandwidth 15), flashes per well 6, read height 4 mm. FITC-dextran concentration was determined in samples from a standard curve generated by serial dilution of FITC-dextran.
RNA processing and RT-PCR
A piece of the jejunum and colon (~5 mm) was incubated in RNAlaterTM (Qiagen) at 4 °C for 48 h and stored at -80 °C until further analysis. For RNA extraction a Tissue-Tearor Homogenizer (Biospec) was used. RNA was extracted using the RNeasy Mini Kit (Qiagen). cDNA synthesis was performed using iScript™ cDNA Synthesis Kit (Bio-Rad) according to manufacturer’s instructions. Expression analysis was performed in duplicate via real-time PCR on a BioRad CFX384 Touch™ Real-Time PCR Detection System using iTaq™ Universal SYBR (Bio-Rad). Expression levels were quantified and normalized to Gapdh expression.
AhR agonist containing supernatant
Similar to previously described (3), to generate AhR agonist containing supernatants, L. reuteri WT or L. reuteri DArAT were grown in MRS broth overnight, harvested by centrifugation, washed with PBS, and 1010 CFU were inoculated into sterile 10 mL of peptone-tryptone water (10 g/L peptone and 10 g/L tryptone, 5 g/L NaCl) supplemented with 0.6 mM L-tryptophan. After a 14 h incubation at 37 °C under anaerobic conditions, bacteria were centrifuged (5000 x g, 10 min), supernatant collected and filter sterilized (0.2 mm pore diameter cellulose acetate filter (VWR)) and stored at -80 °C until further use. For in vitro experiments, supernatants were added to cells at a final concentration of 10% vol/vol in culture media (3).
In vitro naïve CD8 T cell stimulation
Naïve CD8 T cells were purified with EasySep™ Mouse Naïve CD8 T Cell Isolation Kit (STEMCELL, 19858). 5 x 105 naïve CD8 T cells were stimulated with plate coated anti-CD3 (BD Biosciences, 553057) and soluble anti-CD28 (BD Biosciences, 553294) (each 1 μg/mL) in the presence of 10% L. reuteri WT- or 10% L. reuteri DArAT-supernatant. Amount of produced IFNg in the supernatant after 72 h was assessed by ELISA.
Enzyme-linked immunosorbent assay (ELISA) to measure IFNg
In vitro T cell supernatants were used to measure IFNg according to manufacturer’s instructions. Briefly, ELISA plates were coated overnight at 4 °C with 0.5 µg/ml IFNg (BD Pharmingen, 551309). Plates were washed once (0.05% Tween 20 in PBS) and blocked for 1 h with blocking buffer (5% FBS in PBS). Standards and samples were incubated overnight at 4 °C. Plates were washed and incubated with biotin-conjugated IFNg detection AB (0.5 µg/ml, BD Pharmingen, 554410) for 1 h at room temperature (RT). Plates were then washed and incubated with Horseradish Peroxidase-conjugated streptavidin (Jackson ImmunoResearch, 016-030-084) for 10 min followed by washing and development with TMB substrate (Pierce, 34021). Reactions were stopped by the addition of 2N H2SO4, and absorbance was measured at 450 nm on a SpectraMax i3 plate reader (Molecular Devices). IFNg concentration in supernatants was determined from a standard curve generated by serial dilution of IFNg.
AhR activity assay
Luciferase-expressing HT29-Lucia™ AhR reporter cells under the control of Cyp1a1 gene promoter (referred to as “AhR reporter cells”) were purchased from InvivoGen (ht2l-ahr). Cells were cultured in DMEM (Gibco) supplemented with 10% FBS, 1 x Penicillin-Streptomycin-Glutamine (Gibco), 100 µg/mL Normocin (InvivoGen), and 100 µg/mL selective antibiotic Zeocin (InvivoGen). Briefly, 20 µL of sample was incubated with approximately 50,000 AhR reporter cells for 24 h. Following incubation, 20 µL of supernatant was transferred into a 96-Well Clear Bottom Black Microplate (Corning) and 50 µL QUANTI-Luc™ (InvivoGen) was added. Samples were immediately read for luminescence via a SpectraMax® i3x using SoftMax Pro 3.0.7 Software under the following settings: read type endpoint at all wavelengths, integration time 100 ms, read height 2 mm.
AhR activity measurement in tumor homogenate
Pieces of tumor (~ 200 mg) were removed and mashed through 100 µm cell strainers in sterile PBS, followed by centrifugation at 1800 rpm for 5 min to pellet cells. Supernatant was collected and measured for AhR activity using AhR reporter cells.
Bacterial cultures from tumors
Tumors were aseptically removed, weighed, and homogenized via mashing through a 100 µm cell strainer in 2 mL sterile water containing 0.05% NP-40 (Sigma). Bacterial expansion was performed under anaerobic conditions at 37 °C as follows. After a 1 h incubation, 10 mL MRS or Tryptic Soy broth was added to each sample followed by an additional 12 h incubation, after which the expansion and subsequent serial dilutions were plated on MRS or Tryptic Soy agar. Plates were incubated anaerobically for 24 h. Similar as previously described (7), CFUs were then quantified and single colonies were picked for 16S rRNA amplicon sequencing. In Fig. 3D where mice were treated with L. reuteri WT or L. reuteri DArAT additional single colonies were picked for strain specific PCR, where deletion, or lack of deletion, of Lreu23DRAFT_RS05825 was verified using primers 5'-CGACTTGGTGGTCAAAGCGG-3' and 5'-CATTGCTACCCACTTCCTTTACG-3' (3).
AhR activity measurement in translocated bacteria in the tumor
For assessments of AhR activity, MRS-broth-expanded cultures described above were pelleted at 5000 x g for 10 min, resuspended in peptone-tryptone-tryptophan (PTT) media, and cultured anaerobically at 37 °C, shaking at 250 rpm, for 14 h. Bacteria were again pelleted and supernatant was collected and measured for AhR activity.
Bacterial colony identification
Bacterial colonies were identified as described in (7). In brief, grown colonies were picked with sterile pipette tips and stored at -80 °C until analysis. At day of analysis, picked bacterial colonies were thawed at RT, resuspended with 20 µL of sterile water and lysed at 95 °C for 10 min. Samples were subsequently cooled down to 4 °C and then the DNA (2 µl) was used as template DNA in PCR reactions amplifying the 16S rRNA gene using universal bacterial 16S rRNA primers (27F, 5′-AGAGTTTGATCMTGGCTCAG-3′ and 1525R, 5′-AAGGAGGTGATCCAGCC-3′) with reaction conditions: 95 °C for 5 min followed by 35 cycles of 95 °C for 30 sec, 55 °C for 30 sec, 72 °C for 2 min and then 72 °C for 20 min. The amplification product (8 µl) was incubated with 2 µl ExoSAP-ITTM (ThermoFisher) for 37 °C for 15 min, followed by 80 °C for 15 min. As recently described (7), amplicons were sequenced by capillary sequencing, and the resulting sequences were analyzed using BLASTN and the 16S ribosomal RNA sequences NCBI database for species identification.
DNA extraction from intestinal contents and feces
The Fast DNA Stool Mini Kit (Qiagen) was used to extract total DNA from intestinal contents and feces. Quantitative PCR (qPCR) was performed as recently described (8). Briefly, qPCR was performed on a Bio-Rad CFX384 Touch™ Real-Time PCR Detection System using iTaq™ Universal SYBR (Bio-Rad, 1725125) using primers as follows: 16S rRNA-encoding gene (340F, 5’-ACTCCTACGGGAGGCAGCAGT-3’ and 514R, 5’-ATTACCGCGGCTGCTGGC-3’), Lactobacillus reuteri (F, 5’-TTGGAAATGTTCCACAAGAC-3’ and R, 5’-TTGTGAGTTTGGATTGAACC-3), mouse Ifnb1 (F, 5’- CCATCCAAGAGATGCTCCAG-3’ and R, 5’- GTGGAGAGCAGTTGAGGACA-3’). Reactions were run at 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 min and 63 °C for 60 seconds. Expression levels of 16S rRNA-encoding gene and Lactobacillus reuteri were quantified and normalized to Ifnb1 expression.
Dietary experiments
For non-dietary experiments, mice were fed with irradiated standard chow (Prolab, RMH 3000) ad libitum that contains approximately 0.28% tryptophan. For dietary experiments, tryptophan-modified synthetic chows that differ only by their tryptophan content (0.19% or 1.19%; A11022501-02, Research Diets) were fed to mice for the indicated periods of time (ad libitum). Overall food consumption was similar across chows.
Sample preparation of untargeted high-resolution LC-HRMS
Metabolic quenching and polar metabolite pool extraction was performed by adding 360 µL ice cold methanol containing 10 µM 13C1-creatine (Sigma-Aldrich, Boston MA) to 90 µL sample. After 3 min of vortexing, the supernatant was cleared of protein by centrifugation at 16,000 x g. 450 µL cleared supernatant was dried to completion under nitrogen gas and resuspended in 90 µL water. 2 µL of resuspended sample was subjected to online LC-MS analysis. Molar quantities were calculated using a calibration curve using purified kynurenine (Kyn) and indole-3-carboxaldehyde (I3A, Sigma-Aldrich) in a 1:3 series dilution from 25 µM to 11 nM.
Untargeted high-resolution LC-HRMS
Analyses were performed by untargeted LC-HRMS. Briefly, samples were injected via a Thermo Vanquish UHPLC and separated over a reversed phase Phenomenex Kinetix C18 column (2.1 × 150mm, 1.7 μm particle size) maintained at 55 °C. For the 10 min LC gradient, the mobile phase consisted of the following: solvent A (water / 5 mM ammonium formate / 0.1% formic acid) and solvent B (methanol / 0.1% formic acid). The gradient was the following: 0 - 0.3 min 3% B, increase to 30 % B over 0.5 min, continue increasing to 60% B over 1 min, hold at 60 % B for 1.3 min, increase to 95% B over 0.5 min, hold at 95% B for 1.4 min, equilibrate at 3% B for 4.5 min. The Thermo IDX tribrid mass spectrometer was operated in positive ion mode, scanning in ddMS2 mode (2 μscans) from 70 to 800 m/z at 120,000 resolution with an AGC target of 2e5 for full scan, 2e4 for ms2 scans using HCD fragmentation at stepped 15,35,50 collision energies. Source ionization setting was 3.0 kV spray voltage for positive mode. Source gas parameters were 35 sheath gas, 12 auxiliary gas at 320 °C, and 8 sweep gas. Calibration was performed prior to analysis using the PierceTM FlexMix Ion Calibration Solutions (Thermo Fisher Scientific). Integrated peak areas were then extracted manually using Quan Browser (Thermo Fisher Xcalibur ver. 2.7). Purified standards were then purchased and compared in retention time, m/z, along with ms2 fragmentation patterns to validate the identity of peaks.
Statistical analysis
The majority of experiments were repeated at least two times to obtain data for indicated statistical analyses. Mice were allocated to experimental groups on the basis of their genotype and randomized within the given sex- and age-matched group. Given that our mice were inbred and matched for age and sex, we assumed similar variance between the different experimental groups. Statistically significant outliers were excluded from analysis. We did not perform a priori sample size estimation but always used as many mice per group as possible in an attempt to minimize type Ι and type ΙΙ errors. Except mass spectroscopical (LC-HRMS) analysis, investigators were not blinded during experiments and outcome assessment. All experimental and control animals were littermates and none were excluded from the analysis at the time of harvest. All quantitative data are presented as mean ± standard error of the mean (SEM), unless otherwise indicated. Data was analyzed using an unpaired two-tailed Student’s t-test for single comparisons, and one-way or 2-way ANOVA for multiple comparisons. ANOVA analysis was followed by a Sidak’s post-hoc test. Correlations were calculated using the Spearman correlation. Figures and statistical analysis were generated using GraphPad Prism 9 (GraphPad Software). The statistical test used, and P values are indicated in each figure legend. P values of < 0.05 were considered statistically significant. *P < 0.05, **P <0.01, ***P <0.001 and ****P <0.0001.
Data and materials availability: All data are available in the main text or the supplementary materials, except raw CD8 T cell single-cell data, which is available upon request.
Materials and Methods References:
1. D. Davar et al., Phase Ib/II Study of Pembrolizumab and Pegylated-Interferon Alfa-2b in Advanced Melanoma. J Clin Oncol, JCO1800632 (2018).
2. A. Sivan et al., Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084-1089 (2015).
3. L. Cervantes-Barragan et al., Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 357, 806-810 (2017).
4. Y. Hao et al., Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587 e3529 (2021).
5. C. Trapnell et al., The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381-386 (2014).
6. J. Cao et al., The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496-502 (2019).
7. M. Meisel et al., Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580-584 (2018).
8. M. Meisel et al., Interleukin-15 promotes intestinal dysbiosis with butyrate deficiency associated with increased susceptibility to colitis. ISME J 11, 15-30 (2017).
{kind=link}
{kind=link}