Animals
All animal experiments were approved by the first teaching hospital of Tianjin traditional Chinese Medicine Experimental Animal Ethic Committee (Tianjin, China) and were performed under strict supervision. Young male Sprague-Dawley mice, ranging between 45 and 55 g were purchased from the Beijing Vital River Laboratory Animal Technology Co, Ltd, and housed in a temperature(23±2℃) and light (12 hours light/dark cycle)-controlled room with ad libitum access to food and water.
Drugs
YQS mainly consists of sixteen Chinese herbs (Lonicerae japonicae flos, Forsythiae fructus,Schizonepetae spica, Menthae haplocalycis herba, Arctii fructus, Platycodonis radix, Aurantii fructus, Bupleuri radix, Scutellariae radix, Pinelliae rhizome, Arisaema cum bile, Acori tatarinowii rhizome, Curcumae radix, Gastrodiae rhizome,Bombyx batryticatus,Scorpio); Levetiracetam tablets (Capland, B14202068231),o.25g/tablet; β-Actin (abcam, ab79823, dilution 5000 fold); Anti-HMGB1(abcam, ab217274, dilution 5000 fold); Anti-TLR4 (abcam, ab93610, dilution 5000 fold); Goat Anti-Rabbit IgG (ZB-2301, dilution 5000 fold); Goat Anti-MOUSE IgG ( ZB-2305, dilution 5000 fold); Pilocarpine Hydrochloride ( S31556-100mg, LOT:B 12J 10b77879) ; NeuN (E4M5P) Mouse-mAb (CST,94403S,dilution 100 fold); GFAP (GA5) Mouse-mAb (CST,3670S, dilution 300 fold); Mouse Monoclonal CD68/SR-D1 Antibody (NOVUS, NB600-985AF405, dilution 25 fold)
Experimental model and drug administration
All rats were randomly assigned into Control group, Model group, YQS group, LEV group (n=12 each). These groups were further divided into three time points: 7 days, 14 days and 28 days (n=4, each time point). pilocarpine status epileptic model induced as pretest dosage( intraperitoneally injected with 127mg/kg lithium chloride, and 18 hours later, intraperitoneally injected with 200mg/kg pliocarpine ) to observe the frequency and duration time of seizures. When seizures reached the level of Ⅵ or over, lasted for more than 30 minutes seem as successful model. Intraperitoneal injection of diazepam (10mg/kg) to terminate convulsions when convulsions lasting more than 1 hours. All procedures were the same for each group except the control group.
Drug intervention began 30 minutes after the end of convulsions in each group. A total of 146 g crude drug of YQS was concentrated 100 ml, total 250 mg of Levetiracetam was dissolve in 0.9% Nacl to 125 ml. After daily weight measurements, the mice were given average at 1ml/100 g once per day. The Control group and Model group was administrated 0.9% Nacl 1ml/100g by gavage once per day, the YQS group was administrated YQS 1ml/100g by gavage once per day, the LEV group was administrated LEV 1ml/100 g by gavage once per day. Each group at different points were administrated the corresponding drugs or saline interventions for 7 days, 14 days and 28 days respective.
Then brain tissue samples were initiated collection after each time points. The mice were anesthetized by diazepam. Mice were deeply anesthetized and perfused with ice-cold saline, and brain were removed from following decapitation, gently detached and removed the fascia layer and blood of brain tissues. Separated cortical and exposed hippocampus, then were dissected the hippocampus into two parts along the sagittal midline. The left part was flash-frozen in liquid nitrogen, and restored at -70 ℃,which were used for the Western blot. The right part was fixed by 4% paraformaldehyde. Then paraffin sections were prepared by dehydration, transparency, wax dipping and embedding, each brain tissues from continuous coronal sections were sliced for 15 sections with 5 μm thickness, and 50 μm interval between two successive sections, which were used for the Nissl staining, FJB staining, immunoflurosence staining and immunohistochemical staining.
Nissl staining and Fluoro-jade B staining
Paraffin sections were dewaxed routinely and hydrated with gradient ethanol, 1% thionine staining 1ⅹfor 15 min, 95% ethanol 1ⅹto color separation, 100% ethanol 2ⅹfor 1 min to hydration, 100% xylene 2ⅹfor 5 min to transparent. After hydration of gradient ethanol and sealing slices with neutral gum, slices were observed under a microscope. Then Fluoro-jade B staining: Paraffin sections were dewaxed routinely and hydrated with gradient ethanol. 1% NaOH/80% ethanol 2ⅹfor 5 min, 70% ethanol 1ⅹfor 2 min, 0.06% potassium permanganate solution 1ⅹfor 10 min, 0.0004% FJB( room temperature and avoid light) 1ⅹfor 20 min, distilled water washed 3 times, baked at 50-60℃until the slices were completely dry. 100% xylene 1ⅹfor 2 min to transparent and sealing slices with neutral gum. The fluorescence microscope ( excitation wavelength 450-490 nm) was used to observe and collect images.
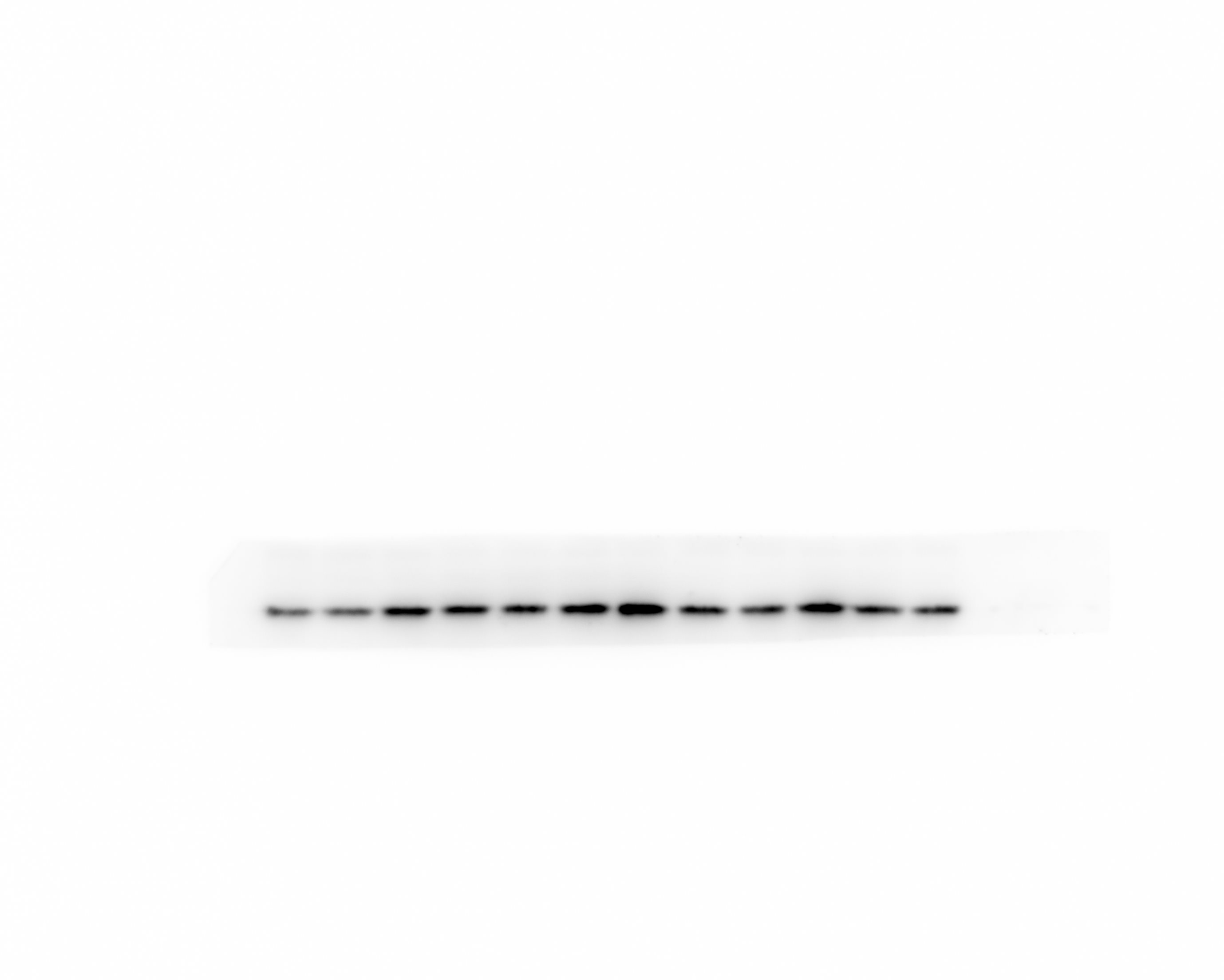
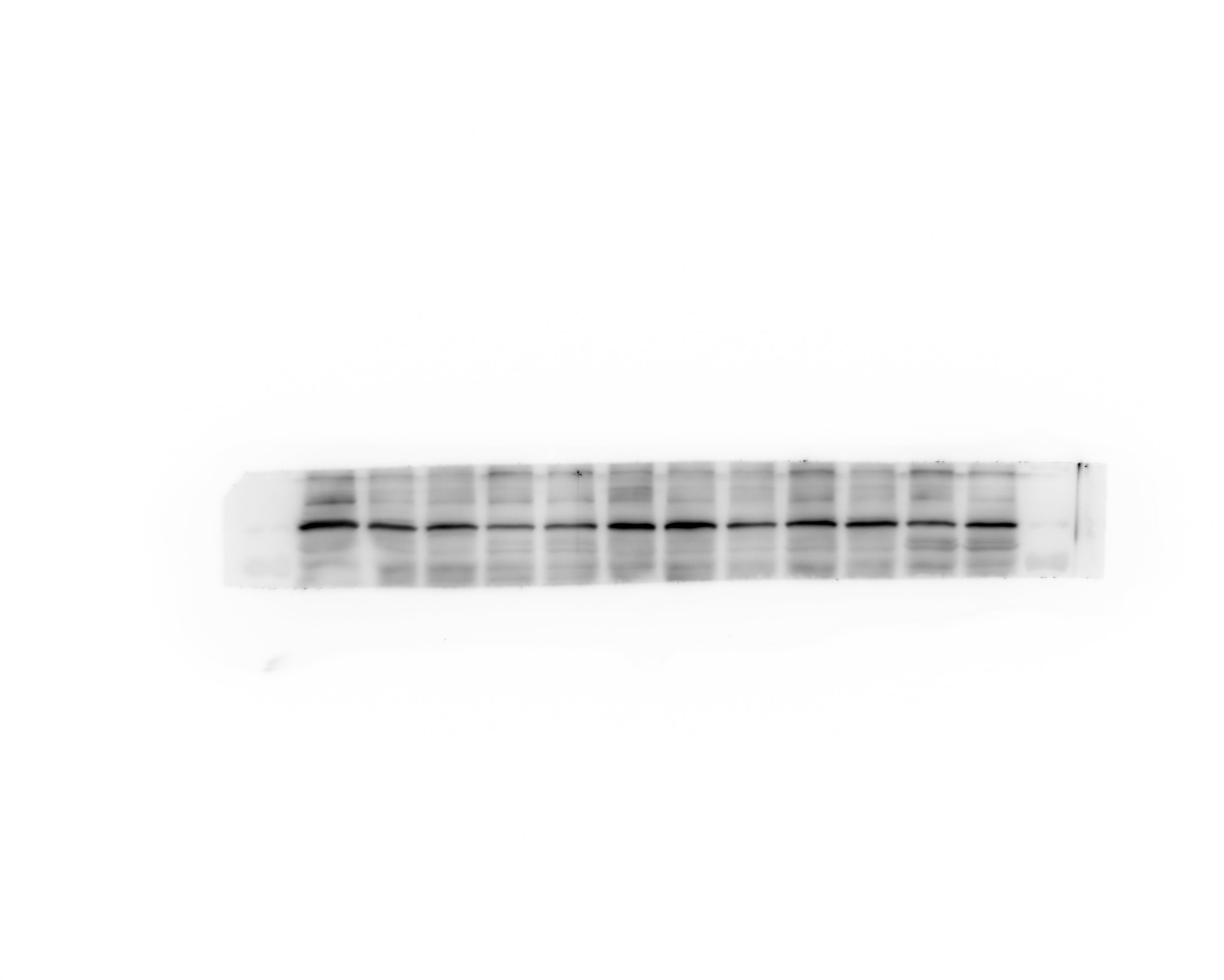
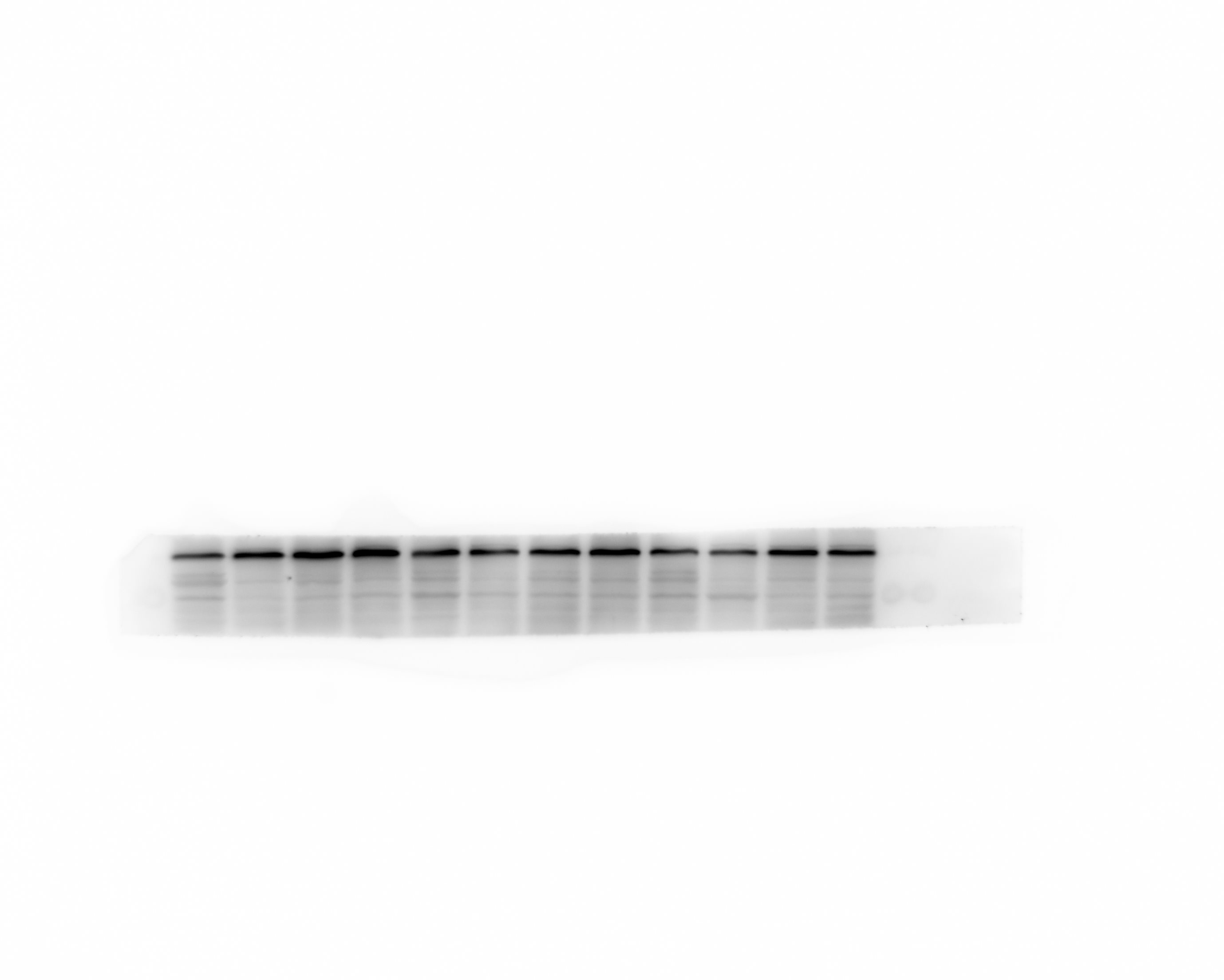
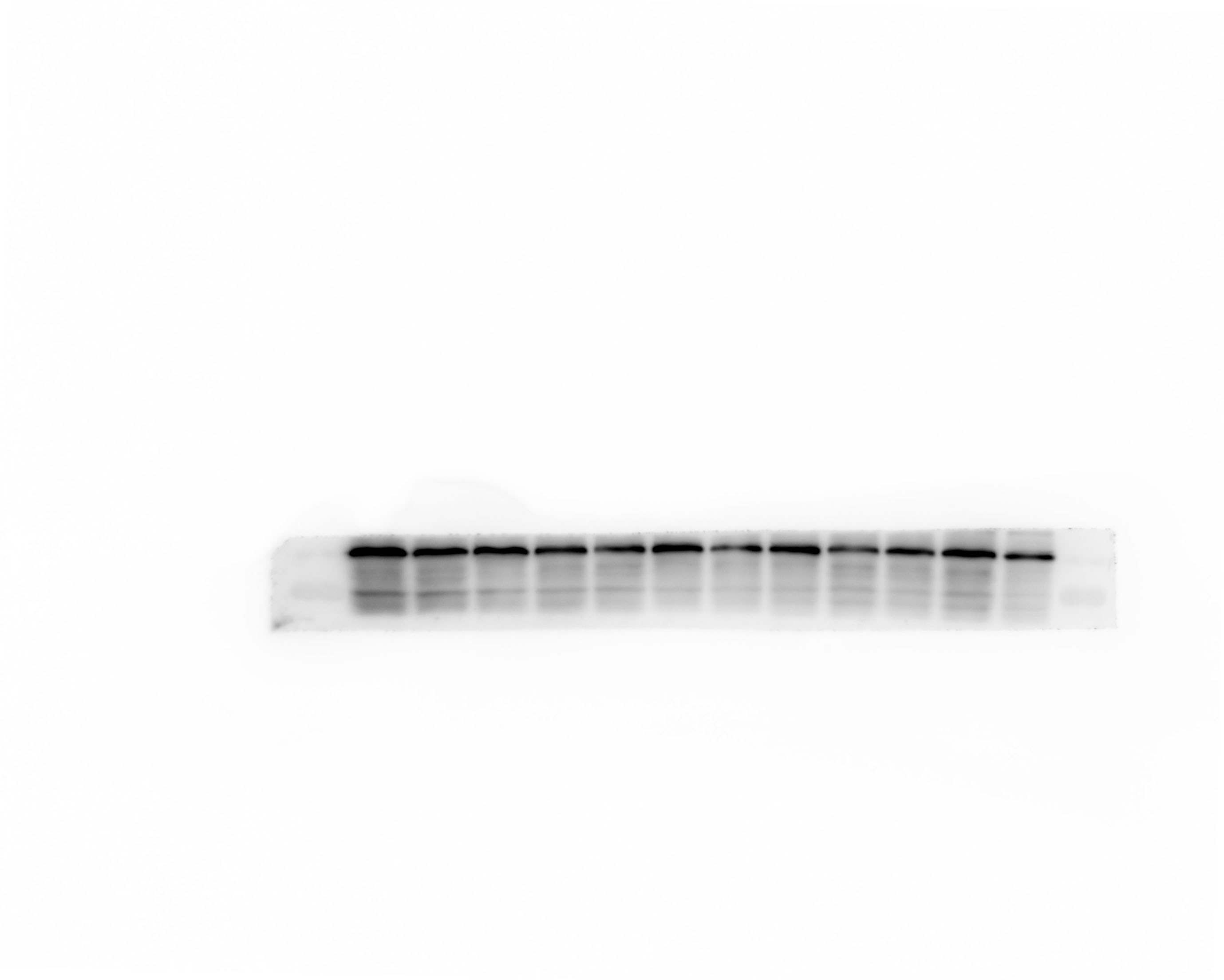
Western blot analysis
Protein were extracted with RIPA lysis buffer (Beyotime Biotechnology, P0013B), and 50μg of total protein was loaded on a gel and separated by sodium dodecyl sulfate polyacrelamide gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membranes and probed with primary antibodies against HGMB1, TLR4, β-actin. Followed by incubation with appropriate Goat Anti-Rabbit IgG (ZB-2301, dilution 5000 fold) secondary antibodies. Immunoblots were visualized using the Western blot detection system. Expression levels were normalized against β-Actin ( abcam, ab79823, dilution 5000 fold).
Immnunohistochemical and Immunofluorescent staining
Paraffin sections were dewaxed routinely and hydrated with gradient ethanol; Antigen repairing: the sections were immersed in citrate buffer( 0.01 M, PH 6.0) and then microwaved for 15 min and cooled to room temperature; rinsed by PBS for 3 times; incubated in 3% hydrogen peroxide at room temperature for 10 min; blocked with 10% goat serum for 30 min; discarded extra serum, added primary antibodies (HMGB1 1:500; TLR4 1:100) and incubated overnight at 4℃;the secondary Goat Anti-Rabbit IgG (ZB-2301, dilution 5000 fold) was added dropwise, incubated at 37℃ for 30 min and then washed with PBS three times for 5 min each time; after washing, streptavidin-HRP were added and incubated at 37℃ for 30 min; DAB substrate kit were added to visualize positive staining and counterstaining with hematoxylin; washed with PBS and differentiated in 1% acid alcohol for 5-10 sec; then dehydrated in gradient ethanol, transparently treated with xylene, sealed with neutral gum, acquisited images with microscope.
Paraffin sections were dewaxed routinely and hydrated with gradient ethanol; Antigen repairing: the sections were immersed in citrate buffer( 0.01 M, PH 6.0) and then microwaved for 15 min and cooled to room temperature; rinsed by PBS for 3 times; incubated in 3% hydrogen peroxide at room temperature for 10 min; blocked with 10% goat serum for 30 min; discarded extra serum, added primary antibodies (anti-HMGB1 1:500; mouse anti-GFAP 1:300; mouse anti-NeuN 1:100;mouse anti-CD86 1:25), incubated overnight at 4℃; the Goat Anti-MOUSE IgG ( ZB-2305, dilution 5000 fold) incubated at 37℃for 45min, protected from light; nuclear staining with DAB incubated at 37℃for5min; sealed with anti-fluorescence quenching sealing tablets and observed under fluorescence microscope.
Statistical analysis
All statistical analyses were performed using SPSS.19 statistical analysis. The results were expressed by mean±standard deviation, statistical differences among the groups were assessed by one-way ANOVA, and post hoc multiple comparisons were performed using Student-Newman-Keuls tests. Values P<0.05 seem as statistically significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}