Experimental designs, animals and animal care
This study was designed with the aim of investigating adult derived-intestinal stem cells and subsequent derivation and cultivation of intestinal organoids in bovine. Hanwoo cattle (Bos Taurus coreanae) (> 24 months old) were used experimentally in this study after receiving approval from the Institutional Animal Care and Use Committee (IACUC) of the National Institute of Animal Science (NIAS-2019-366), Korea. All procedures, including sample collection and handling, followed the standard operating protocols of the Animal Biotechnology Division at the National Institute of Animal Science, Korea.
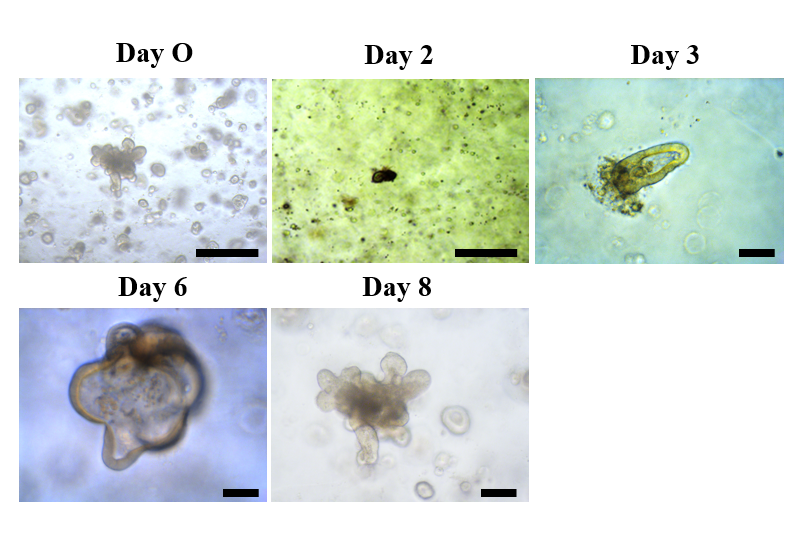
Isolation of intestinal crypts and three-dimensional (3D) cultivation
Different jejunum fragments from the small intestine were obtained from Hanwoo cows and collected in washing buffer containing ice-cold phosphate-buffered saline (PBS) containing
1% penicillin/streptomycin (Sigma-Aldrich, NY, USA). The fragments were opened longitudinally and washed thoroughly with washing buffer to remove the debris. Mucosal and submucosal layers were gently scraped off using a glass slide. The remaining muscle layer was collected into a 50-ml tube containing 30 ml of washing buffer after being cut into 3- to 5-mm pieces, repeatedly washed by shaking vigorously and centrifuged at 300 g until the supernatant was clear. The collected pellet was resuspended in 25 ml of Cell Disassociation Solution (StemCell Technologies, Vancouver, Canada) and incubated at room temperature for 40 min on a rocker to release the crypts. Crypts were collected after pipetting and centrifugation at 300 g for 5 min. The pellet was resuspended in 1 ml of intestinal human organoid medium (StemCell Technologies), and the crypts were counted under an inverted microscope. The seeding mix composed of medium with 100-150 crypts, and Matrigel in a 1:1 ratio was prepared and then placed in the middle of a 24-well plate. The cells were returned to an incubator to polymerize the Matrigel, and after 20 min, 1 ml of organoid growth medium was gently added to each well.
Passage and cryopreservation of bovine intestinal organoids
Bovine intestinal organoids were subjected to passage approximately once a week upon maturation. Briefly, the medium was gently aspirated and rinsed with ice-cold PBS without disturbing the organoid dome. To harvest the organoids, a 10× volume of enzyme-free cell disassociation buffer (1 ml) was added to a Matrigel dome (100 μl) in each well and incubated for 10 min in an incubator. Organoids were dislodged by gentle pipetting and collected by centrifugation at 300 g for 5 min. The pellet was resuspended in the desired amount of medium and Matrigel in a 1:1 ratio, and each well (140-150 organoids) was distributed into three parts in subsequent passages and seeded in 24-well plates. For cryopreservation, organoids were resuspended in preserving solution composed of 90% medium and 10% dimethyl sulfoxide (DMSO) (Sigma-Aldrich), stored at -80°C for 24 hr and transferred to a liquid nitrogen tank for long-term storage.
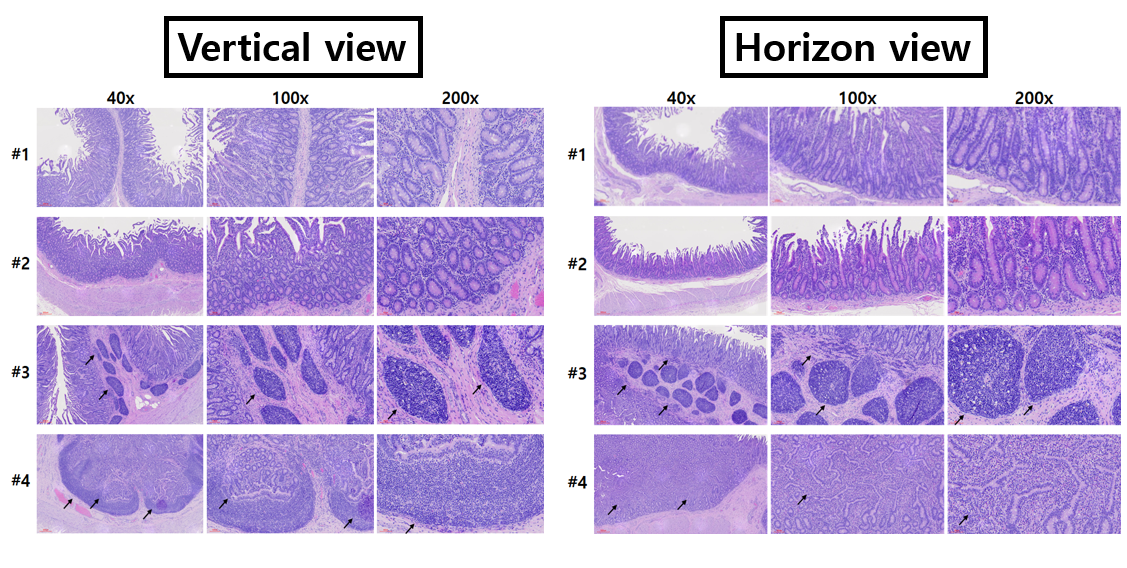
Histology and immunohistochemistry
The small intestine segments were fixed in 10% neutral-buffered formalin (Sigma-Aldrich) after strong washing with ice-cold PBS. The segments were subsequently embedded in a paraffin block, and the paraffin-embedded intestinal tissue was vertically and horizontally sectioned at a thickness of 3-5 μm. The sections were then deparaffinized in xylene, rehydrated with water via a graded alcohol series, and processed prior to haematoxylin and eosin (Merck, Darmstadt, Germany) staining. In the immunohistochemical analysis, the sections were permeabilized with 0.1% Triton X-100 in PBS for 5 min and incubated with 0.1% normal goat serum for 1 h to block nonspecific binding after antigen retrieval by boiling the sections in a sodium citrate buffer solution. The samples were incubated with appropriate dilutions of primary antibodies at 4°C overnight. The antibodies used in this study are shown in Table 1. After washing, the samples were reacted with anti-mouse and anti-rabbit secondary antibodies coupled to Alexa Fluor-488 and Alexa Fluor-594 (Molecular Probes/Life Technologies, Waltham, MA, USA), respectively, for 1 h at room temperature. These fluorescent samples were counterstained with diamidino-2-phenylindole (DAPI). The images were captured using an Olympus X100 confocal microscope (Olympus, Tokyo, Japan).
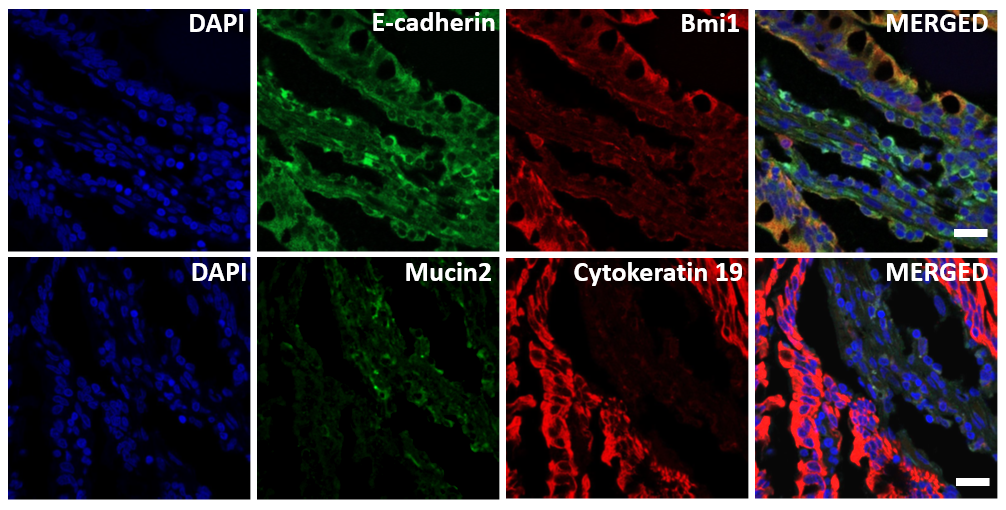
Immunocytochemistry
The organoids were maintained in 24-well plates until maturation. The fixation media was aspirated in the wells, and the organoids were washed thoroughly with cold PBS and incubated in neutrally buffered 4% paraformaldehyde (Sigma-Aldrich) for 30 min at room temperature. Then, the organoids were permeabilized in buffer containing 0.5% Triton X-100 (Sigma-Aldrich) in PBS for 30 min at room temperature. The blocking step was performed using 3% bovine serum albumin (BSA) in PBS for 1 h at room temperature. The organoids were thoroughly rinsed with PBS and incubated overnight at 4 °C with the appropriate primary antibodies, as shown in Table 1, at their appropriate dilutions. The marker gene expression was detected by incubating the samples with corresponding secondary antibodies coupled to AlexaFluor-488 and AlexaFluor-594 (Molecular Probes/Life Technologies) for 1 h at room temperature. These fluorescent samples were counterstained with diamidino-2-phenylindole (DAPI) and mounted on glass slides using ProLong Gold antifade (Life Technologies, Waltham, MA, USA) mounting medium. The images were captured under an Olympus X100 confocal microscope (Olympus).
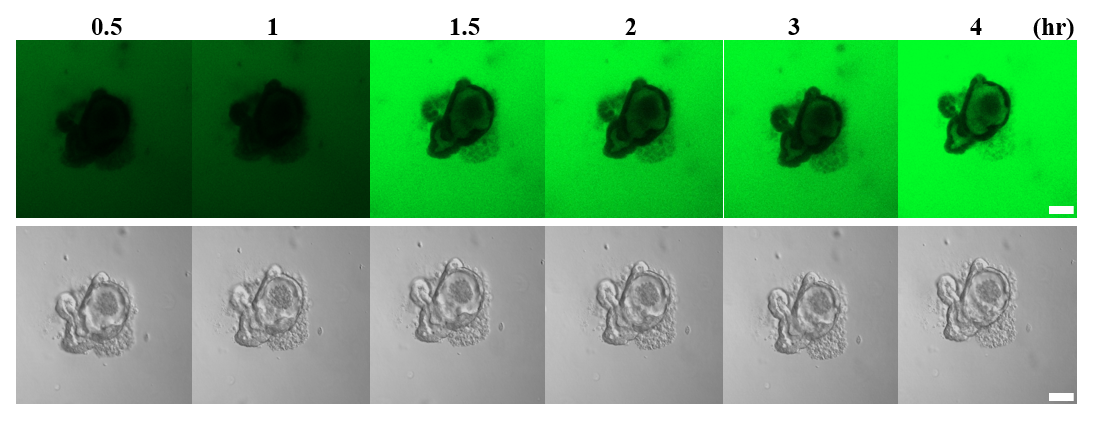
Epithelial barrier permeability assay using FITC-dextran
Epithelial barrier function was tested by diluting powdered Fluorescein isothiocyanate (FITC)-dextran (4 and 40 kDa) (Sigma-Aldrich) in nuclease-free water, which resulted in a 1 mg/ml working solution. The organoids were placed in 24-well plates and allowed to grow until fully developed into crypt and villi structures. Then, 25 ng/ml FITC-dextran was added to each well, and the plate was incubated under normal growth conditions. The permeability was observed using luminal absorption and recorded for more than 180 min at 30-min intervals under a Leica CTR6000 fluorescence microscope (Leica, Wentzler, Germany).
RNA isolation
Total RNA for prepared samples including intestinal organoids was isolated using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) as described previously [14, 15]. RNA quality was assessed by an Agilent 2100 bioanalyzer using an RNA 6000 Nano Chip (Agilent Technologies, Amstelveen, The Netherlands), and RNA quantification was performed using an ND 2000 Spectrophotometer (Thermo Inc., DE, USA).
Library preparation and sequencing
For control and test RNAs, library construction was performed using QuantSeq 3’ mRNA-Seq. Library Prep Kit (Lexogen, Inc., Austria) according to the manufacturer’s instructions. In brief, each 500 ng of total RNA was prepared, an oligo-dT primer containing an Illumina-compatible sequence at its 5’ end was hybridized to the RNA, and reverse transcription was performed. After degradation of the RNA template, second strand synthesis was initiated by a random primer containing an Illumina-compatible linker sequence at its 5` end. The double-stranded library was purified using magnetic beads to remove all reaction components. The library was amplified to add the complete adapter sequences required for cluster generation. The finished library is purified from PCR components. High-throughput sequencing was performed as single-end 75 sequencing on a NextSeq 500 (Illumina, Inc., USA).
Data analysis
QuantSeq 3` mRNA-Seq. reads were aligned using Bowtie2 [16]. Bowtie2 indices were generated from either the genome assembly sequence or the representative transcript sequences for alignment to the genome and transcriptome. The alignment file was used for assembling transcripts, estimating their abundances and detecting differential expression of genes. Differentially expressed genes were determined based on counts from unique and multiple alignments using the coverage command in Bedtools [17]. The RC (read count) data were processed based on the quantile normalization method using EdgeR within R (R Development Core Team, 2016) using Bioconductor [18]. Gene classification was based on searches performed in the DAVID (http://david.abcc.ncifcrf.gov/) and Medline databases (http://www.ncbi.nlm.nih.gov/).
Data availability
QuantSeq 3’ mRNA-Seq data sets are available via the following accession code in the Gene Expression Omnibus database (GEO): GSE163425
Statistical analysis
All data are expressed as the means ± standard error of the mean (SEM) from three independent experiments. Significant differences between groups were analysed by one-way ANOVA or Student’s t-test. A P value less than and equal to 0.05 indicated statistical significance (*P value ≤ 0.05).
{kind=link}
{kind=link}
{kind=link}
{kind=link}