In this study we have reported a clear difference in faecal microbial composition and predicted function between adult horses of the same breed and gender, but different levels of domestication; ranging from no human intervention to high human intervention (i.e., Feral – Semi-feral – Domesticated). The overall structure of the microbiome in the study participants was similar to that previously described for horses, with the most abundant phyla being Firmicutes and Bacteroidetes [45,21,46,47,48,22,25]. There were clear differences in global composition between the groups according to unsupervised beta diversity analysis. This was supported by supervised multivariate analysis using CCA, in which the differences between the three groups were significant (p= 0.001). Clustering of individuals according to PCoA (Fig 2b) showed that the greatest difference in microbial beta diversity was between Domesticated and Feral groups, and the Semi-feral animals spanned between them, indicating step-wise differences in the composition of the faecal microbiota in response to increasing levels of domestication. The predicted functional changes mirrored those in the micorbiome data, with a network analysis revealing the least correlation between the Feral and Domesticated groups. To our knowledge, this is the first time a step-wise response to domestication level has been described in the equine GI microbiota.
Based on previous studies of the equine microbiome [49], and given that all animals were clinically healthy at the time of sampling, the differences in management between groups which were likely to be responsible for the observed differences were diet [31,19, 50,51,17,52], drug use [53,48,54,57], level of GI parasites [48,54,55] and, potentially, handling stress [56]. We attempted to dissect these factors by recording the dietary differences, drug use and GI helminths burden (by proxy of FEC) in each group at the time of sampling. It being spring, the main difference in diet between groups was the level of access to managed ryegrass pasture versus rough moorland grazing; although the Semi-Feral and Domesticated groups had also received supplementary hay and concentrate (in the case of the latter) over the winter. None of the animals in the study had been treated with an antibiotic, nor anthelmintic in the last 8 weeks, and previous studies have shown that anthelmintic use alone has small and transient effects on the GI microbiota [48,54,57,53]. Therefore, we assumed that there were no direct effects of anti-infective drugs at the time of sampling. However, evidently the different parasite treatment protocols have led to different FEC profiles between groups.
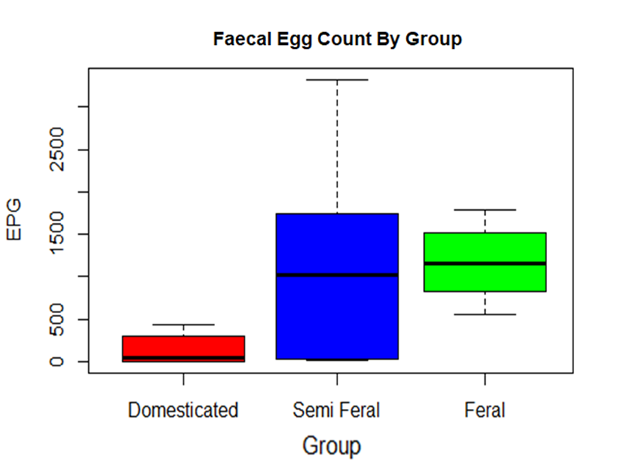
Strongyle FECs were significantly lower in the Domesticated group (Additional File 4), which is expected as these animals were tested and treated more regularly for these parasites. The average FEC was similar between the Feral and Semi-feral group, however there was much greater variation in parasite burden between animals in the Semi-Feral group (Additional Table 2; Additional File 4). Of note, the ponies in this group were wormed only once per year, and they were kept in a relatively restricted area which would allow for the build-up of parasites on the pasture. Therefore, it is likely that these animals were exposed to high helminth infection intensity and, consequently, parasite burdens would fluctuate widely between treatments. Over the course of the year, the diet of the Semi-feral herd would also be expected to fluctuate more than the other two groups as they were grazed on different pasture types and had sporadic supplementation with hay. Both dietary changes and acute helminth infection have been shown to cause alterations to microbial composition and alpha diversity in horses [31,19,50,51,17,52,48,54,55] and, interestingly, there was also a much higher variation in the microbial alpha diversity in the Semi-feral group when compared to the Feral and Domesticated group (Additional File 2). Furthermore, the predicted functional analysis for the Semi-feral group included increases in KEGG pathways for cancers, metabolic diseases and immune system diseases, all of which appear to represent an unhealthier phenotype. This may be an indication that a lack of consistency in diet and parasite management on a small holding may be more disruptive to gut microbiota than either a Feral or a Domesticated system with relatively stable management. Indeed, parasite treatment and changes in diet have both been found to be precede GI disease in horses [16,58,59,60].
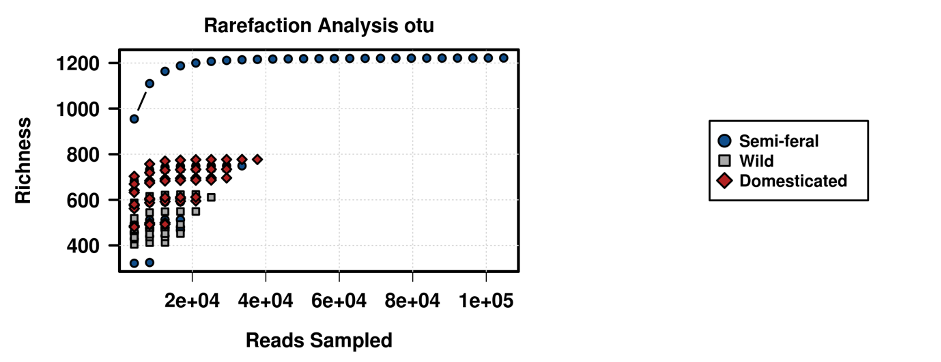
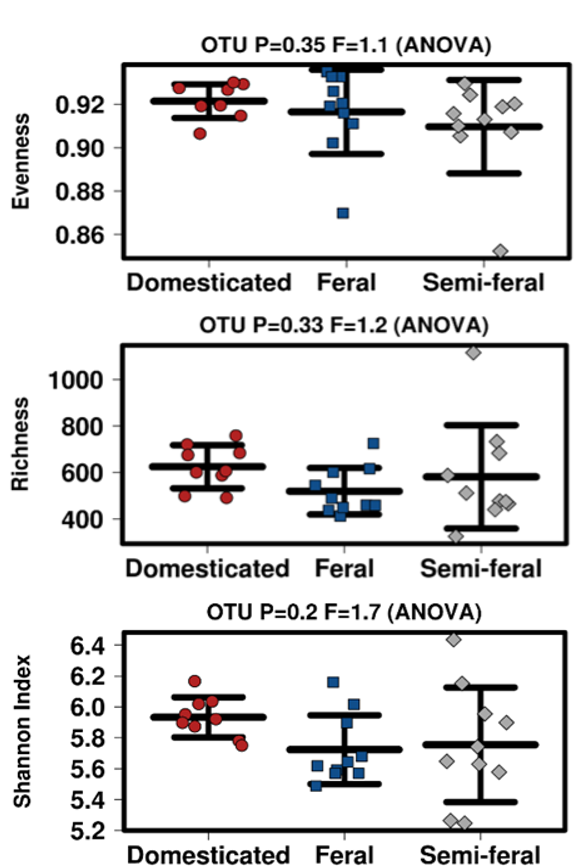
In contrast to some previous studies comparing wild and domesticated populations [4] the average alpha diversity between groups in this in this study did not differ significantly (P=0.2). Notably, reduced diversity has been associated with domestication and GI disease in many species including horses, and based on this data it is tempting to infer that low diversity is a contributing factor in diseases resulting from domestication [61,62,63,64,65,66]. However, an increasing body of evidence, including our data, calls this assumption into question, as no significant differences in alpha diversity between equid groups were reported [31,32,33]. Indeed, it may be that a high variation in alpha diversity (as was seen in the Semi-Feral herd), or a sudden drop in alpha diversity within a given population, is a better indication of GI health than the diversity metric itself.
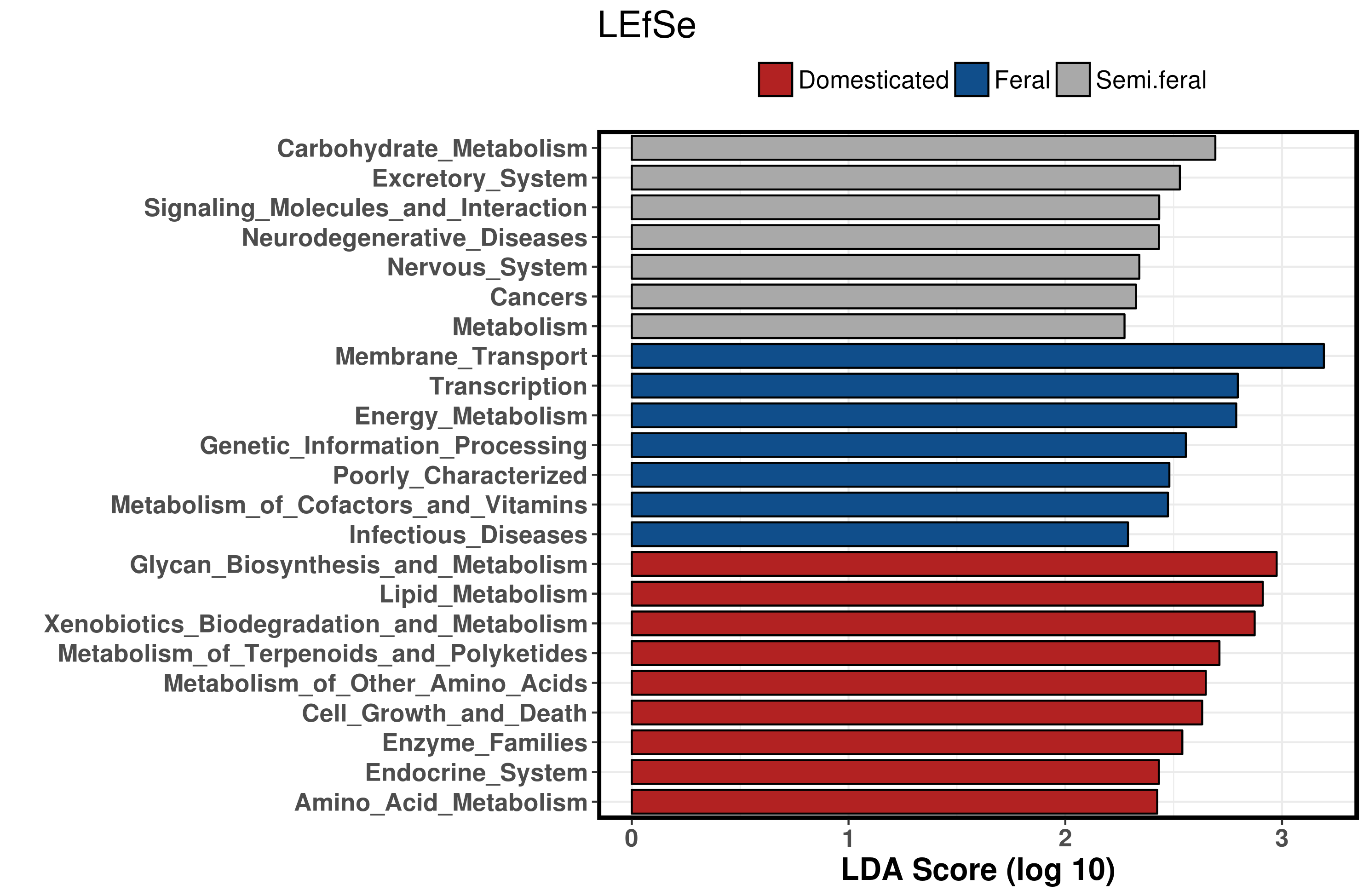
Overall, taxonomic analysis using a combination of LEfSe and network analysis corroborated the findings of multivariate analyses, demonstrating clear step-wise changes in the abundance of many bacterial taxa between the Domesticated, Semi-Feral and Feral groups (Figs 2,3,4). With respect to the Domesticated group, the taxa Kirimatiellaeota (Kiritimatiellae), Tenericutes (Anaeroplasma, Mycoplasma), Alphaproteobacteria (Rhizobales), Deltproteobacteria (Desulfovibrio), Synergistes (Pyramidobacter), Actinomycetales (Mycobacterium), Negativicutes (Acidaminococcaceae), Optituales and Lentispherae (Victivallaceae) showed a step-wise decrease in relative abundance from Domesticated, to Semi-Feral, to Feral. Of these taxa Kirimatiellaeota, Lentispherae, Alphaproteobacteria and Tenericutes were also negatively correlated with FEC which, given the domesticated group had the lowest FEC, may indicate that high levels of these bacteria were related to low parasite burden. On the other hand, this may actually be a false correlation, since there was a significantly lower FEC in the Domesticated group; indeed, we were not able to find examples of negative relationships between these bacterial taxa and helminth infection in the literature.
Notably, members of the Classes Alpha- and Delta-proteobacteria were significantly higher in the apparently healthy Domesticated ponies. Higher levels of the Class Alphaproteobacteria were largely attributable to increases in the order Rhizobales; whereas an increase in Class Deltaproteobacteria was attributable to an increased abundance of genus Desulfovibrio. In general, elevated levels of Proteobacteria in horses have been linked to increased starch intake [19,50,67]. The domesticated group had the highest level of access to ryegrass pasture, which has a higher starch level than rough moorland grasses [68,69,70]; therefore, the increase in Proteobacteria seen in the Domesticated group may be linked to dietary starch levels. The long-term effects of concentrate supplementation over the winter may also have contributed to this finding, although there are no studies evaluating the residual effects of starch in horses to confirm this hypothesis. Similar increases in Proteobacteria in horses have also been associated with intestinal inflammation, colic and equine grass sickness [22,71,72,73]. In particular, Weese et al (2015) showed that Proteobacteria increased in horses in the days prior to presenting with colon torsion, suggesting that bacteria belonging to this phylum may play a role in precipitating disease. Therefore, increases in Proteobacteria associated with dietary starch intake, such as seen here, may be a risk factor for the development of GI disease. Other changes in taxa which linked to diet in the Domesticated group included a relative increase in Negativicutes (Acidaminobacteriaceae) abundance. These bacteria use amino-acids as their sole source for growth, and so their increase is consistent with a higher protein diet, consistent with a diet of ryegrass versus rough moorland grasses [68,69,74]; furthermore, these findings would explain the functional prediction of increased amino acid metabolism from PICRUST analysis.
In the Domesticated group there were also relative increases in bacterial taxa (e.g. Tenericutes, Synergistes and Actinomycetales) which have been described either in horses, or other species, as pathobionts. For example, the increase in Tenericutes was largely attributable to increases in the abundance of Anaeroplasma and Mycoplasma. These bacteria are gram-negative, anaerobic, and found in mammalian gut microbiota [75,76]. They are considered a pathobiont and are known to increase in response to GI inflammation in mice and humans; for example, Mycoplasma are implicated in the aetiology of Chrohn’s disease in humans [76,77]. Mycoplasma are also a cause of respiratory disease in horses [78,79,80]. The elevated abundance of these taxa may therefore indicate an ‘unhealthy’ gut microbial profile, however there is currently no data in horses to prove this hypothesis.
With regards to the Feral group, Chlorflexi (Anaerolineaceae), Actinobacteria (Coriobacteriia; Eggerthellaceae) Atopobiaceae, Coriobacteriaceae), Euryarchaeota (Methanobrevibacter) and Clostridiales (Lachnospiracaea, Shuttlesworthia) showed step-wise decreases in abundance from Domesticated > Semi-feral > Feral. Chlorflexi (Anaerolineacae), Actinobacteria (Coriobacteriiia), and Lachnospiraceae (Shuttlesworthia) were also positively correlated with FEC. Coriobacteriales have been found in previous work to increase with helminth infection [80]. Actinobacteria were also increased in helminth infections in cats [82] and mice [83], and they have been shown to increase in response to helminth infection with Trichuris suis infection in pigs alongside dietary inulin supplementation [84]. In the latter case, these changes were linked to an anti-inflammatory Th2 immune response. Hence, it is plausible that some bacteria belonging to these taxa have a genuine link with helminth infection, which may explain their presence at higher levels in the Feral group.
As for the Domesticated group, many of the other taxa which were associated with the Feral group had strong links to diet. In particular, Euryarchaeota (genus Methanobrevibacter) was the bacterial taxa most strongly associated to this group. Methanobrevibacter spp facilitate the fermentation rate and colonic energy production in the form of SCFA by removing H2 from bacterial fermentation [84], which is important to utilise energy from fibrous diets. High levels of Euryarchaeota are associated with high fibre diets in horses [86,46]; and have also been linked to wild horses in studies comparing wild and domestic populations [32]. Therefore, the association of these taxa with the Feral group in this study most likely reflects the relative high fibre content of the rough moorland diet of the Feral population. Anaerolinaceae are anaerobic fermenters producing methanogens, synergistically linked with members of Euryarcheota [87], such as Methanobrevobacter. Furthermore, Coriobacteriaceae have been shown to increase in abundance proportionately to increasing concentration of cellobiose supplementation in horses [88]. Cellobiose forms when cellulose is partially hydrolyzed by the enzyme cellulase [89], suggesting that an increase in these bacteria could be a downstream effect of a high fibre diet. Lachnospiraceae are also associated with the breakdown of cellulose and hemicellulose [90] which may indicate that these bacterial taxa were increased as a result of the high level of fibre in a rough moorland diet. Increased abundance of Lachnospiraceae due to diet may, in turn, have positive health effects for the horse as increased levels of this family have been reported in the faecal microbiome of healthy horses when compared with animals with colitis [22] and diarrhoea [91]. The predicted functional analysis performed in this study supports the role of the microbiota of Feral group in fibre digestion, with KEGG pathway for energy metabolism being significantly increased in the Feral group compared with the other two groups.
{kind=link}
{kind=link}
{kind=link}
{kind=link}