We envisaged that the three diverse reaction manifolds observed by TAS are governed by alternative orbital interactions. For instance, the interactions within pathway iii differ from pathways i and ii lacking the π orbital overlap of the indole with the half-filled np orbital of the excited ketone. Indeed, it is well established that for EDA-complexes alternative p-p interactions are relevant16. Importantly, diverse orbital interactions may lead to diverse spatial orientations of the reagents and finally alter the stereoselectivity of the process. For this reason, we decided to gain insights into the diverse molecular orientations by using DFT methods.
The Exciplex vs. the EDA-complex Pathway. Computational Insights
Aiming at developing a novel stereodivergent process, we focused our attention on the exciplex-based pathway ii and the EDA-complex-based pathway iii, with 1b and 2b as reactants.
Under diluted conditions ([2b] = 0.01 M in Acetone), no EDA complex is detected, meaning that under these conditions the only active pathway is the exciplex-based pathway ii. Conversely, when more concentrated conditions ([2b] = 0.1 M in Acetone) are used the EDA-complex pathway iii starts to be competitive27.
To gain insight into the two diverse arrangements involved in the PET-based pathways (exciplex vs. EDA complex), we employed DFT calculations at the (U)M06-2X/6-311 + + G(d,p)/ IEFPCM(acetone) level of theory (Fig. 3)17. This functional is a well-established method to describe charge transfer complexes both in singlet and triplet states28, 29.
Excited-state (T 1 ) – Exciplex arrangement, pathway ii (Fig. 3a). Upon direct excitation of the ketone 2b, the corresponding T1 state is generated. At this juncture, the T1 interacts with the ground state 1b10. In this case, the oxygen atom of the T1 2b and the C2 position of 1b are in close proximity (Fig. 3a, np-π interaction, 2.56 Å). Interestingly, the spin density map of the computed exciplex is almost equally distributed between the indole and the ketone (Fig. 3, 1.03 and 0.97 respectively). On the one hand, a large dipole moment is observed upon excitation (µ = 15.6 D). This altered charge distribution indicates a remarkable CT character as reflected by natural population analysis. In fact, a total of 0.90 charge unit is transferred from indole to the ketone, corroborating the PET-based pathway ii, where the radical ion-pair has been detected experimentally by LFP measurements.
Ground-state (S 0 ) – EDA-complex arrangement, pathway iii (Fig. 3b). As experimentally observed, when concentrated conditions are used ([2b] = 0.1 M in Ace) an EDA-complex is generated between the reagents. This complex is held together through a π-π stacking interaction between the electron-rich aromatic ring of indole 1b and the electron-poor aromatic ring of the ketone 2b (Fig. 3, interaction ~ 3.6 Å and corroborated by computed UV-Vis spectra in Fig. S.I.8.)30, 31. Additionally, the analysis of the frontier molecular orbitals of the EDA-complex reveals that the HOMO is located on the indole moiety while the LUMO is distributed along the ketone resulting in a dipole moment (µ) of 4.8 D (Fig. 3). To better elucidate the photoinduced electron transfer observed after irradiation of the EDA complex, we performed vertical excitation calculations employing a TD-DFT method. This technique allowed us to analyze the corresponding excited state involved in the PET process. For the S1 excited-state, a HOMO-LUMO contribution of 83.5% has been computed (3.66 eV), showing a remarkable charge transfer character (Fig. 3i, µ = 16.2 D). Indeed, natural population analysis of S1 indicates that upon vertical excitation of the EDA complex a substantial charge (0.79) is transferred from the indole moiety 1b to the ketone 2b, which resembles the experimentally observed radical ion-pair system.
Exciplex vs. EDA complex. The impact of the light source on the diastereodifferenciation process
Based on the different geometries and orbital interactions obtained by DFT calculations, it is possible to represent the two alternative reaction manifolds by using a simplified potential energy surface diagram (Fig. 4)31. It is worth reminding, that the spatial arrangements of the molecules are retained and directly transferred to the final oxetane product due to the concerted-character of PET-based PB reactions11, 12. For instance, when the reaction mixture is irradiated at 405 nm, the CT-band of the EDA complex is directly excited (Fig. 4 left, red lines). This irradiation leads to the formation of a radical IP with a preferred π−π interaction. On the other hand, 2b can be directly excited at 370 nm. In this case the np-π interaction will govern the spatial arrangement of the IP (Fig. 4 right, green lines). Thus, a light stereodifferentiation processes can occur when switching the irradiation source from UV- to visible-light.
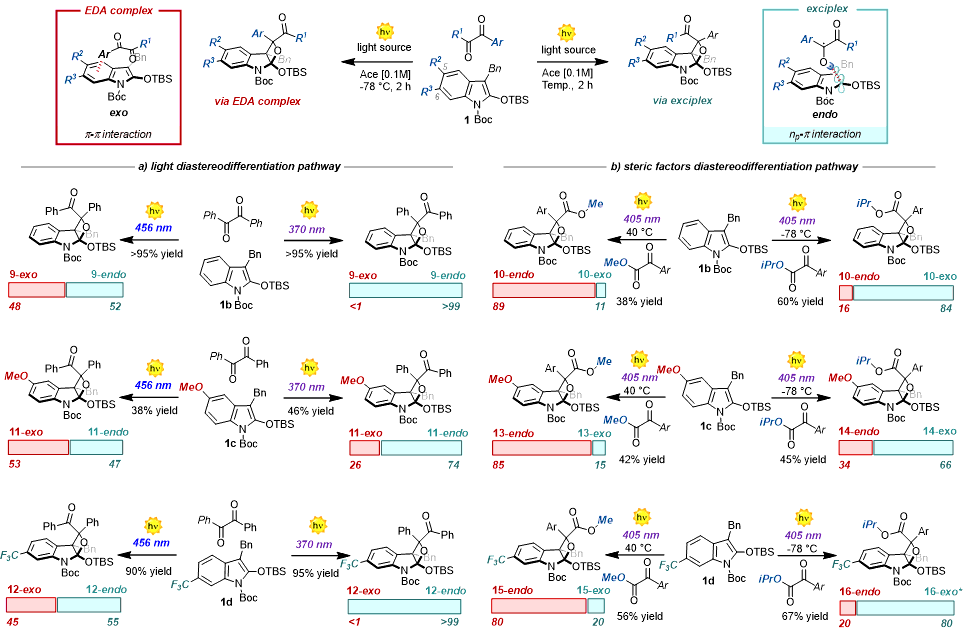
As per the DFT calculations, there are two alternative sets of interactions, which do not result in any product diversification when using symmetric benzophenones. These are crucial when moving to prochiral carbonyls. We therefore evaluated benzil 7, which is a class of synthetically relevant prochiral carbonyls used in several polar and radical reactions (Fig. 5)32, 33. We hypothesized that the endo:exo selectivity24 of the reaction would depend on the competition between the two alternative reaction manifolds (exciplex vs. EDA complex). The conventional pure-triplet mechanism (pathway i) was ruled out by the LFP experiments of 7 with 1b (see Section F in SI), which instead confirmed the photogeneration of the CT state, in agreement with the favorable ΔGPET (-5.8 kcal·mol-1). Thus, by altering the irradiation wavelength and the parameters that influence the EDA complex formation, the endo/exo selectivity can be deliberately altered in a diastereodivergent light-driven process (see Section E and F in SI). Specifically, 8-endo will be the preferred product in an exciplex-based mechanism, while 8-exo will be favored following an EDA complex pathway (Fig. 5, left). Remarkably, we observed the formation of 8-endo as a single diastereoisomer when running the reaction at 370 nm (> 99:<1 dr, 370 nm Fig. 5b). The endo selectivity results from the direct excitation of the free ketone 7, participating in an exciplex with 1b.19 As hypothesized, when moving to less energetic 405 and 456 nm irradiation wavelengths, the exo selectivity increases, with the dr moving to 77:23 (456 nm Fig. 5b). This is in line with the activity of the excited EDA complex between benzil 7 and the indole 1b, whose presence was confirmed by the UV-Vis absorption spectra (Fig. 5c). Increasing the concentration from 0.01 to 0.1 M favors the EDA complex pathway, resulting in a dr of 93:7 and 68:32 upon irradiation at 370 and 405 nm, respectively. No significant variation was observed at 456 nm, indicating that 64:36 dr is the limit value under these reaction conditions. Following previous experimental observations, we reasoned that performing the reaction under reduced temperature should inhibit the interconversion between the two reaction manifolds, thus magnifying the observed light stereodifferentiation process.35. Indeed, when running the reaction at -78°C, the dr moved from > 99:<1 at 370 nm to 52:48 at 456 nm. This dr variation is the highest ever registered for PB light-diastereodifferentiation processes, and it confirmed our initial working hyphothesis36 − 38. Furthermore, these experimental data reveal that the choice of the light is crucial. Reproducibility issues in diastereoselective PB processes involving heterocyclic systems can be now rationalized and possibly solved by selecting the appropriate irradiation wavelength39. As a control experiment, we performed a reaction in an aromatic solvent, which interferes with the π−π interactions in the EDA complex. Accordingly, when using C6D6 as solvent, we observed reduced activity of the EDA-complex pathway, resulting in a dr no lower than 80:20 at 465 nm (Fig. 5b, right column).
In addition to the experimental data, we used DFT and TD-DFT calculations to compute the two different geometries for the EDA complex between ketone 7 and indole 1b (see section K in SI). Notably, the excited state (S2, HOMO-LUMO contribution = 98.4%) of this complex in the exo configuration has a larger dipole moment (µ = 18.6 D) than in the endo geometry (µ = 15.4 D). This indicates a substantial charge-transfer character and is in accordance with the experimental trend.
Spectroscopical characterization of the exciplex intermediate, pathway ii (Fig. 6).
In order to confirm the key reactivity of an exciplex between substrates 1b and 7, we performed transient absorption spectrometry (TAS) measurements at a fs timescale. These experiments allowed investigating the dynamics of the systems within the first nanosecond after photoexcitation with a time resolution of about 150 fs (see SI, section F). We compared the photophysics of the single benzil 7 in the absence and in the presence and of indole 1b (Fig. 6a and b, respectively). All the experiments were conducted under the reaction conditions ([1b] = [7] = 0.1M in Ace).
For 7, the excitation at 400 nm results in the formation of positive excited state absorption features (ESA). At early time, we recorded a broad absorption band centered at about 560 nm, which gradually evolved into a sharper feature with a maximum at about 525 nm. While the literature contains few reports of the ultrafast dynamics of 7, which are typically performed at longer time resolution, it is reasonable to assign the early time signal at 560 nm to the ESA from the instantaneously photoexcited S1 state (S1®Sn transition)41, 43. This signal quickly decays (0.3 ps) while the band at 525 nm rises with the same time constant. Once formed, this band decays over a longer timeframe than the explored 600 ps. This behavior is in agreement with a fast S1®T1 ISC that moves the population from S1 to T1 on a sub-ps timescale, followed by the long-lived T1®Tn absorption at 525 nm44.
The TAS spectra of the mixture 7-1b (Fig. 6.b) are qualitatively similar to the TAS spectra of 7. This is not surprising, given that the excitation at 400 nm preferentially excites 7 and therefore its spectral features are expected to prevail in the mixture response. However, the dynamic behavior is significantly different, with the ESA band at 525 nm decaying much quicker in the mixture than in 7 (Fig. 6c-d). The time trace of 7 alone could be fitted with a bi-exponential model characterized by a fast rise (trise=0.3 ps) assigned to the S1®T1 ISC the and a slow tdecay (> 500ps) assigned to all the relaxation processes from the T1 state to the S0 ground state. Interestingly, an additional relaxation pathway is observed for the triplet state in the presence of 1b, with a time constant of about 50 ps, which is associated with the formation of triplet exciplex species as depicted in Fig. 6e. Indeed, the formation of the triplet state is known to lead to a planarization of the benzil molecule,44 which favors the exciplex formation, as confirmed by our TD-DFT calculations (see section K in SI). We can exclude that the 50 ps time constant corresponds to the direct formation of a 7-1b CT state. This is because TAS experiments performed on the 7-1b mixture confirmed that this species gives rise to a characteristic ESA signal at significantly longer wavelengths (ca 600 nm) and has much longer dynamics.
Adding to previous experimental evidence and supported by DFT calculations, the findings support the existence of an exciplex between substrates 1b and 7 under these experimental conditions, while offering a rare report of the activity of this elusive and fleeting intermediate.
Exciplex vs. EDA complex. The impact of the electronic and steric factors on the diastereodifferenciation process
Having verified the key activity of the exciplex under the light-driven stereodifferentiation processes, we next investigated how steric and electronic parameters can be used to rationally control the stereodifferentiation events. Specifically, we selected α-ketoesters 9 as a molecular probe45 to monitor the impact of steric and electronic parameters on the reaction outcome, while keeping constant a visible-light source set at 405 nm (Fig. 8). Interestingly, the steric repulsion between the R group and the substituent at the C-2 of the indole 1b (OTBS) will favor a carbonyl perpendicular orientation in the exciplex, leading to 10-exo (Fig. 8, exciplex box). However, the 10-endo selectivity will be preferred when favoring a p-p interaction between the aryl group of 9 and the aromatic indole core (Fig. 8, EDA-complex box).
Evaluation of the electronic factors (Fig. 8b). We began by evaluating the impact of the electronic factors on the reaction outcome. First, we selected various α-ketoesters 9 recording the corresponding UV-Vis spectra in the presence of 1b, thus assessing their ability to generate an EDA complex. In the case of α-ketoester with Ar = 3,5-CF3Ph, we observed a red-shifted CT band (see Fig. 5a and Section G in SI). Additionally, the highly negative ΔGPET = − 17.1 kcal·mol− 1 measured was consistent with the formation and activity of an EDA complex. No spectral variations were registered for the other α-ketoesters. We next examined the experimental outcome for all the substates under a 405 nm irradiation (Fig. 5b).
Using a visible-light source was crucial because it enabled the activity of the exciplex and the EDA complex pathways (see section G in SI). This meant that the electronic and steric parameters were the key features in governing the selectivity of these photoreactions46. In fact, both the EDA complex and free α-ketoesters absorb light under these conditions. Only the PB reaction with the electron-deficient 3,5-CF3Ph α-ketoester was highly endo-selective (84:16 dr, Fig. 8b). The other α-ketoesters showed no ground-state association with 1b, while maintaining a high level of exergonicity (ΔGPET in the range of -6.0 to -9.5 kcal·mol− 1, Fig. 8b) and resulting in similar dr of about 70:30.11, 24
We depicted the differences in reactivity by plotting the obtained experimental dr against the Ered of 9 and highlighting the different behavior of the 3,5-CF3Ph α-ketoester (Fig. 8b, right).
Evaluation of the steric factors (Fig. 8c). We then investigated the role of the steric factors, tuning the R group within 9. The Charton analysis allowed us to compare the sensitivities (ψ) of the two different manifolds (EDA complex vs exciplex).47 When increasing the R size (from Me to iPr), the endo/exo selectivity was reversed, passing from 74:26 to 43:57 dr for Ar = Ph (exciplex manifold), and from 84:16 to 45:55 dr for Ar = 3,5-CF3Ph (EDA complex manifold). We rationalize this trend as reflecting an increased steric repulsion between the R group and the OTBS, favoring the perpendicular approach. Nevertheless, the EDA complex manifold experiences a 1.5 increased sensitivity (ψ = -1.51 vs ψ = -2.23 for the exciplex and EDA manifolds, respectively), in agreement with a greater dependence of the parallel approach (π−π interactions) towards steric variations. Finally, we sought to use these findings to deliberately alter the diastereoselective outcome of the PB process by manipulating the steric factors.47 We selected the 3,5-CF3Ph α-ketoester because it is the only substrate that formed an EDA complex with 1b (ψ = -2.23). We observed an initial dr of 84:16 in favor of the endo product. The endo selectivity increased slightly at 40°C (89:11).48, 37 When passing from R = Me to R = iPr, the endo:exo selectivity was reversed with a dr of 45:55 and further magnified to 16:84 when lowering the temperature (-78°C). We thus isolated and characterized the elusive exo-isomer of oxetane 10 in synthetically useful isolated yield (58%). These results demonstrate that simple structural variations (Me versus iPr) in the starting material can dramatically alter the stereochemical outcome of light-driven processes. The importance of the proper light-source selection is also key. No variations were observed when running the reaction at 370 nm. Moreover, these results indicate that overlooked minor modifications of a substrate can have a tremendous impact on the reaction outcome of [2 + 2] photocycloadditions.
Following the rational approach used for the previous reaction partners, computational analysis was performed (see section K in SI). We computed the EDA complexes and the corresponding vertical excitations between the α-ketoester (9, R = Me) and indole 1b. Remarkably, the corresponding excited state (S1, HOMO-LUMO contribution = 94.4%) of the EDA complex in the endo configuration (major diastereoisomer) has a larger charge-transfer character (µ = 15.8 D) than the exo geometry (µ = 14.3 D). This trend is in agreement with previous data on the benzil-indole couple. Moreover, similarly to the 1b-2b exciplex, the complex in the triplet state between 1b and 9 (R = Me) possesses a large dipole moment (m = 15.9 D), as reflected by the charge transferred (0.95) from indole to the ketone, which supports the PET-based pathway.
{kind=link}