MSCs and tumor cell lines
Human umbilical cord MSCs (hUC-MSCs) were obtained from the Biotherapy Center of Harbin Medical University Cancer Hospital and prepared following a well-established protocol[28]. The human umbilical cords were donated by women who had underwent eutocia. Informed consents was obtained from the subjects’ families and the study was approved by the Harbin Medical University Cancer Hospital Ethics Review Board. Purity was confirmed by flow cytometry (CD73+, CD90+, CD105+, and CD146+, CD31-, CD34-, CD45-, HLA-DR-). These MSCs can differentiate into osteoblasts, chondroblasts and adipocytes in vitro. MesenCult™ MSC Basal Medium (Stem Cell) was used to culture MSCs without serum in a GMP-grade condition. The 4th passage was used for the following experiments. The differentiation of MSCs to adipocytes, osteocytes and chondrocytes was tested by using StemPro® and an adipogenesis kit (cat no.A10070-01, Gibco), osteogenesis kit (cat no.A10072-01, Gibco) and chondrogenesis differentiation kit (cat no.A10071-01, Gibco). Afterward, staining with Oil Red Oranse, Alizarin Red S, and Alcian Blue was also performed to detect adipocytes, osteocytes and chondrocytes, respectively.
The normal colonic mucosa cell line NCM460 was purchased from the Shanghai Cell Bank. The human HCT116 colorectal carcinoma (ATCC CCL‐247™), SW480 adenocarcinoma carcinoma (ATCC CCL‐228™), Caco2 adenocarcinoma carc carcinoma (ATCC HTB-37™), Lovo colorectal adenocarcinoma (ATCC CCL‐229™), HT29 adenocarcinoma adenocarcinoma (ATCC HTB-38™) and SW620 colorectal adenocarcinoma [American Type Culture Collection (ATCC)® CCL‐227™] cell lines were purchased from the ATCC. With the exception of Caco2, the other cell lines were grown in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic solution in a 5% CO2 incubator. Caco2 cells were cultured in DMEM supplemented with 20% FBS.
Isolation and purification of research-grade exosomes
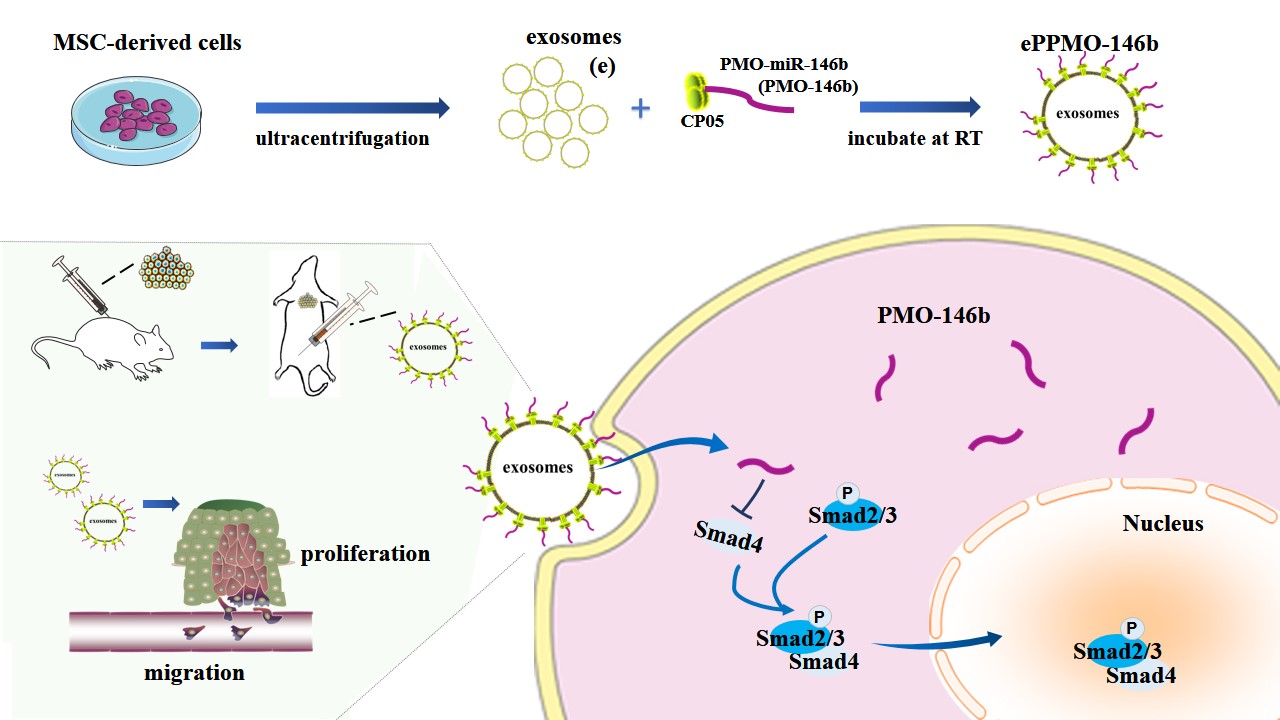
hUC-MSC-derived exosomes were purified by ultracentrifugation. Supernatants were collected from MSCs cultured as monolayers in serum-free medium and were subsequently subjected to centrifugation at 3,000 × g for 30 min. The supernatants were then filtered using 0.2-μm filters, and the pellet was recovered and subsequently ultracentrifuged (Beckman, USA) at 100,000 × g using a SW32 Ti rotor for 3 h. The supernatants were aspirated and the resulting pellet was suspended again in PBS and again centrifuged at 100,000 × g for 1 h. The supernatants were aspirated and the pellet was recovered in 2 mL PBS and stored ar -80℃ until use.
Transmission electron microscopy (TEM)
The exosome suspension was diluted with PBS at a 1:1 ratio, and 10 μL of this solution was dropped onto formvar-carbon-coated grids and blotted with filter papers after sedation for 1 min. Then, 10 μL of 3% phosphotungstic acid was dropped onto the exosome area for 1 min. After the excess staining buffer was removed with filter papers, the grid was left to air-dry for 5 min. Exosome morphologies were visualized using a high-resolution transmission electron microscope (Hitachi HT7700, Japan) at 80 kV.
Nano flow cytometry (NanoFCM) for exosome size and concentration analysis
Flow NanoAnalyzer model type N30 (NanoFCM Inc., China) was used to determine the exosome size distribution and granular concentration according to the manufacturer’s instructions. Briefly, the isolated exosomes were diluted with PBS at a 1:100 ratio. The Silica Nanospheres Cocktail (S16M-Exo, NanoFCM Inc., China) was employed to construct a calibration curve regarding particle size and side scattering intensity. Using this calibration curve, the side scattering intensity of every exosome was converted into the corresponding vesicle size.
Flow cytometry analysis of exosome-bound beads
Exosomes from MSCs were isolated as described above and resuspended in 200 μL PBS. Aldehyde/sulfate latex beads (10 μL, A37304, Life Technologies, USA) were added to the solution and mixed using a benchtop rotator for 15 min at room temperature. PBS (600 μL) was then added to the solution and mixed overnight at 4°C. The mixture was then spun down at 8,000 × g for 1 min. The precipitate was then resuspended in 100 μL of 10% BSA in PBS and mixed for 45 min at room temperature. The mixture was spun down at 8,000 × g for 1 min, and the supernatant was aspirated. Exosome-bound beads (the pellet) were then resuspended in 20 μL PBS and immunolabeled for CD63, CD73, CD90 or an isotype control. The beads were incubated with 1 μL anti-CD63 antibody (Catalog #12-0639-42, eBioscience, USA) or 1 μL anti-CD73 (Material Number 550257, BD Biosciences, USA) or 1 μL anti-CD90 antibody (Material Number 555596, BD Biosciences, USA) or 1 μL Mouse IgG1, κ isotype control antibody (Material Number 555749, BD Biosciences, USA) in a final volume of 20 μL and mixed at room temperature for 30 min in the dark. The mixture was then centrifuged at 8,000 × g for 1 min, the supernatant was aspirated, and the pellet was resuspended in 200 μL PBS with 2% BSA. The expression of exosomal markers (CD63) and mesenchymal markers (CD73 and CD90) was analyzed using flow cytometry (BD FACSAria II analyzer, USA). Data were analyzed using FlowJo software (TreeStar Inc.). The flow cytometry experiment was repeated two times independently using the same exosome preparation.
Synthesis of antisense oligomers
The morpholino oligos (also known as PMOs) were synthesized by Gene Tools LLC. (USA), including negative controls (NC) and anti-miR-146b-5p ASO (PMO-146b), all of which were modified on the 3' ends-biotin and 5’ ends-primary amine. The purity of PMO was determined to be 95% using reverse-phase HPLC and MALDI TOF mass spectrometry. PMO sequences are listed in Supporting Information Table S1. The peptide CRHSQMTVTSRL (CP05) was conjugated with PMO-146b (PPMO-146b) as previously described[25, 29].
Flow cytometry and fluorescence microscopy
To measure the binding affinity of candidate peptides to exosomes (e), 1.32×1010 e were preincubated with biotin-labeled CP05-anti-miR-146b‐5p ASO (PPMO-146b) or biotin-labeled anti-miR‐146b‐5p ASO (PMO-146b; as a control) overnight at 4°C (PPMO-146b or PMO-146b solution (10 µg/µL) resolved in PBS at 1 mM), followed by washing with PBS for five times in 1.5-mL ultracentrifuge tubes and filtration with Amicon Ultra-0.5 Centrifuge Filter (Millipore) to remove unbound peptides (14,000 g, 10 min). Subsequently, the exosome-PMO complexes, ePPMO-146b and ePMO-146b were incubated with 4-µm aldehyde/sulfate latex beads for 15 min at room temperature under rotation and washed with PBS for three times (8,000 g, 1 min each time). The segregated complexes were then incubated with 3% BSA-DPBS for 30 min on a rotator. After washing with PBS for 3 times (8,000 g, 1 min), streptevidin-PE (1:500, diluted in 3% BSA blocking buffer, Catalog Number 12-4317, eBioscience) was added to the complexes and incubated for 30 min at room temperature in the dark. After washing 3 times with PBS, the recovered beads were observed with a conventional fluorescence microscopy (Zeiss, Germany) or subjected to flow cytometry (FACSCalibur, BD). Uncoated beads were used as negative controls for gating. We used the separation index (SI) metric that was defined by Theodoraki et. al, which takes into account both the difference in mean fluorescent intensity (MFI) between PPMO-146b and PMO-146b SI = \(\frac{\left({MFI}_{PPMO-146b}-{MFI}_{PMO}-146\text{b} \right)}{\sqrt{\frac{{{SD}_{PPMO-146b}}^{2}+{{SD}_{PMO-146b}}^{2}}{2}}}\) capture beads, and the average of their distributions[30].
Uptake of PKH67-labeled ePPMO-146b by SW620 cells
To determine whether SW620 CRC cells could take up targeted exosomes from hUC-MSCs, the PKH67 Green Fluorescent Cell Linker Kit (Thermofisher) was used to label exosomes, according to the manufacturer’s protocol. Briefly, MSC-derived exosomes, ePMO-146b-biotin and ePPMO-146b-biotin were diluted and resuspended in sterile PBS to a final concentration of 5×107 particles. Diluent C (500 μL; 2×PKH67 solution) was added to 2-µL PKH67 dye (PKH67GL, Sigma), and 500 μL exosomes were mixed with 500-μL PKH67 solution to allow internalization. Subsequently, incubate the exosomes/dye mixture for 1-5 min with periodic mixing, and then the staining was stopped by adding an equal volume of serum or 1% BSA-PBS. Then, the unbound PKH67 was removed by using Amicon Ultra-0.5 Centrifuge Filter (Millipore) at 14,000 × g for 10 min.
SW620 cells were seeded in 12-well plates at a density of 1×105 cells/well and were incubated in a complete medium for 12 h. Subsequently, the plates were rinsed twice with PBS and fixed with 4% paraformaldehyde solution at 4℃. The plates were then rinsed again three times using PBS. After rewashing, 10 µM unlabeled avidin (S888, Invitrogen, USA) was added to link biotinylated PMO on the membrane surface for 30 min at 4℃.
Next, the cells were permeabilized with 0.1% Triton X-100 for 5 min. PBS with 3% BSA was used to incubate with the cells for 2 h to block nonspecific binding. Then Streptavidin R PE (SNN1007, Invitrogen, USA) was utilized for the detection of PMO-labeled biotinylated anti-miR-146b-5p ASO in presence or absence of CP05 peptides at room temperature for 1 h. Cells incubated with native exosomes and PBS with the same volume as the suspension of PMO-146b-loaded exosomes served as controls.. Finally, the cells were washed three times with precooled PBS and were mounted with DAPI-containing mounting media (H-1800, Vector Labs). Images were captured under a fluorescence microscope (Zeiss, Germany).
Quantitative real-time PCR
Total RNA was extracted from seven cell lines with TRIzol reagent (Invitrogen). The cDNAs were produced from the RNA samples with PrimeScriptTM 1st Strand cDNA Synthesis Kit (No. 6110A, TaKaRa, Japan). Human U6 snRNA was used as an endogenous control for data normalization. Real-time PCR was performed on the LightCycler®96 system (Roche, USA) using TB GreenTM Premix Ex TaqTM (No. RR820L, Takara, Japan). Relative miRNA expression was determined using the Ct method. All experiments were performed at least three times. Oligonucleotides were synthesized by Integrated DNA Technologies (Sangon Biotech, China) and the primer sequences were designed as previously described[19]. The primer sequences are listed in Table S1.
Cell viability analysis with a by CCK8 assay
SW620, Caco2 and Lovo cells were seeded in 96-well plates at a density of 5-8×104 cells/well. Once the cells reached an approximately about 80% confluency, they were starved overnight and cocultured with different concentrations of exosomes (2×109, 2×1010 and 2×1011 particles/mL) or an equal volume of PBS as a control. In another experiment, the cells were exposed to exosomes, ePNC, ePPMO-146b or PBS. Cell growth was analyzed 48 h after the exosome treatment. CCK8 solution of 10 μL (Beyotime, China) was added to each well and the solutions were incubated at 37 °C for 4 h, then the optical density was detected at a wavelength of 450 nm by a microplate reader (Thermofisher, USA). The data shown are representative of at least three independent experiments.
Wound‐healing assay
SW620 and Caco2 cells were seeded onto a six‐well plate at a density of 5×105 cells/mL until they reached full confluency. The cells were cultured for 24 h in the presence of exosomes, ePNC or ePPMO-146b. Then each well was scratched by using a 200-μL pipette tube to create 2 linear regions that were devoid of cells, and medium without FBS was added. PBS was used as an additional negative control. Photographs were captured by a digital single lens reflex camera (Canon) at 0 and 24 h, respectively. The migration area of the cells was measured by ImageJ software.
Transwell migration assay
SW620 and Caco2 cells (4-5×104 cells/well) were seeded on upper chambers (6.5 mm Transwell® with 8.0-μm pore polycarbonate membrane insert; Corning) in serum‐free medium with 0.1% BSA. A total of 600 μL medium containing 10% FBS was added to the lower chambers. After incubation for 24 h, nonmigrating and noninvading cells were gently removed with a cotton swab. The cells were then fixed with methanol for 30 min and stained with 0.1% crystal violet for another 20 min. The area of dyed pores was then calculated under a microscope at a magnification of ×200. Three views were selected randomly for photography and analysis. Each measurement was repeated three times.
Western blot analysis
Total proteins from cells or exosomes were extracted at 4°C by using RIPA Lysis Buffer (Beyotime, China) with a protease inhibitor (Roche, USA), and were then vortexed every 5-10 min for 30 min. Subsequently, the lysates were spun down at 14,000 × g for 20 min to remove any debris and the supernatant was collected. Exosomes (20 μg) and protein samples (70 μg per lane) were loaded onto 10% SDS-PAGE gels and transferred onto PVDF membranes (Millipore, USA) for 90 min. For immunodetection, the membranes were incubated with the following primary antibodies at 4°C overnight: CD63 (ab134045), CD81 (ab109201), TSG101 (ab125011), E-cadherin (ab76055), vimentin (ab92547), and N-cadherin (ab76011). All primary antibodies were purchased from Abcam (Cambridge, UK).The next morning, the membranes were washed with TBS-T three times for 10 min each time. After incubation with secondary antibody at room temperature for 1 h, the membranes were washed with TBS-T and then incubated with luminol substrate solution (Transgene, China) for 1 min. Images were collected with a chemiluminescence imaging system (Proteinsimple). All experiments were performed at least three times.
In vivo experiments
Our protocol for animal use in this project was approved by the Animal Experimentation and Ethics Committee of Harbin Medical University Cancer Hospital and Peking University Shenzhen Hospital, and all animal experiments were carried out according to the Guide for the Care and Use of Laboratory Animals, and in strict accordance with the People’s Republic of China Legislation Regarding the Use and Care of Laboratory Animals.
Antitumor efficacy of ePPMO-146b in tumor-xenografted nude mice
Adult female athymic BALB/c nude mice (15-20 g) that were 8 weeks old, were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. [Certificate no. 110011211106810132 SCXK (Jing) 2021-0006]. The animals were housed in a controlled environment at 23±2°C under a 12-h dark/light cycle with free access to irradiated food and sterile water. To ensure the number of tumor-bearing mice, 10 female BALB/c nude mice were subcutaneously injected with Lovo cells in 200-μL of PBS (2×106 cells). One week later, the tumor-bearing mice were approximately 90 mm3 in size. The tumor-bearing mice were randomly divided into ePNC group and ePPMO-146b group (n=5) on the basis of their tumor volume. EPNC solution and ePPMO-146b solution (5.0×1010 particles/kg in 10 μL) were injected into the tumor-bearing mice via a peritoneal injection. The mice were treated twice a week for 24 days and tumor size was measured once or twice a week for 24 days with a vernier caliper. Tumor volumes were calculated according to the following formula: Volume (mm3) = length × (width)2/2. The mice were killed at 25 days post-injection of ePNC and ePPMO-146b. The inhibitory rate of tumor volume was calculated as IR (%) = (Vt–V0)/V0 × 100%, where V0 is the tumor volume of the epNC group mice and Vt is the tumor volume of the ePPMO-146b group. The inhibitory rate of tumor weight was calculated as IR (%) = (Wt–W0)/Wt × 100%, where Wt and W0 represent the tumor weights of the ePNC and ePPMO-146b-treated groups, respectively.
Biodistribution of systemical ePPMO-146b
DiR-labeled exosomes were prepared according to the literature [31]. Purified exosomes (5.0×109 particles/mL) were incubated with 1 μM fluorescent lipophilic tracer DiR (D12731, Invitrogen, USA) for 5 min at 37℃ prior to 4℃ for another 15 min. Unbound dye was removed by centrifugation at 100,000 g overnight by means of an SW41 Ti rotor (Backman), and then the pellet was washed twice with precooled PBS. The obtained DiR-Exos were resuspended in precooled PBS, and the particle concentration was measured by nanoFCM. DiR-Exos were diluted in PBS to achieve a particle concentration of 8.0×109/mL.
Female healthy male C57BL/6N mice (Animal Center of the 2nd Affiliated Hospital of Harbin Medical University) or C57BL/6 nude mice aged 8 weeks were used. Freshly purified DiR-labeled exosomes and naked exosomes were injected intraperitoneally (i.p.) in the C57BL/6N mice. The biodistribution of DiR-labelled exosomes was examined using the same dosage mentioned above, and the particle count was measured with nanoFCM and the samples were diluted to 200 μL. To analyze DiR exosome distribution, IVIS Spectrum (Perkin Elmer) was performed. Live mice (isoflurane sedated) were imaged after 3 h posttreatment. For ex vivo imaging, the mice were sacrificed after 6 h postinjection and the liver, heart, lung, kidney, spleen, intestine and reproductive organs were collected. The live mice or the harvested organs were imaged for 1-2 seconds (at an excitation wavelength: 640 nm, and an emission wavelength of 710 nm).
DiR-ePPMO-146b was intraperitoneally injected in the tumor-bearing C57BL/6 nude mice. At 6 h postinjection (5.0×1010 particles/kg), organs were harvested and prepared as described above.
Statistical analysis
Statistical comparisons among multiple groups were determined by one-way analysis of variance (ANOVA) with Dunnett’s post-hoc test. A probability value of 0.05 was considered to be significant. The results are expressed as the mean ± SEM.
{kind=link}