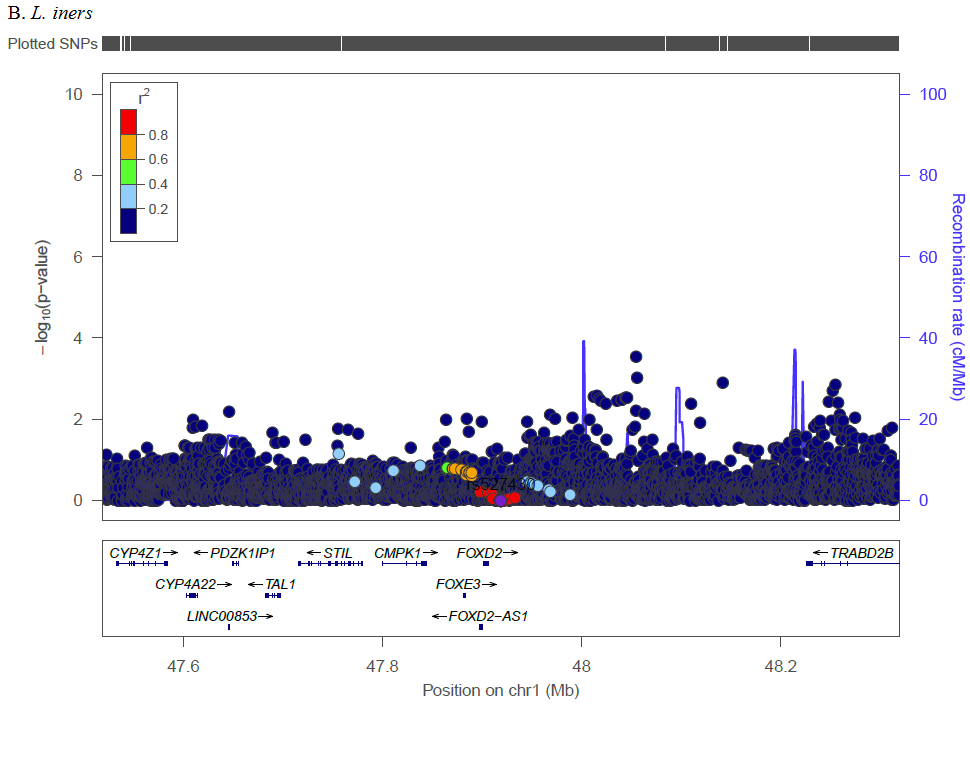
To our knowledge, this is the first GWAS of vaginal microbiome traits. In native Kenyan women, we identified novel genomic loci and biological pathways with biological relevance to host immunity, cell signaling, and infection, supporting the role of host genetics in inter-individual variability in vaginal microbiome traits.
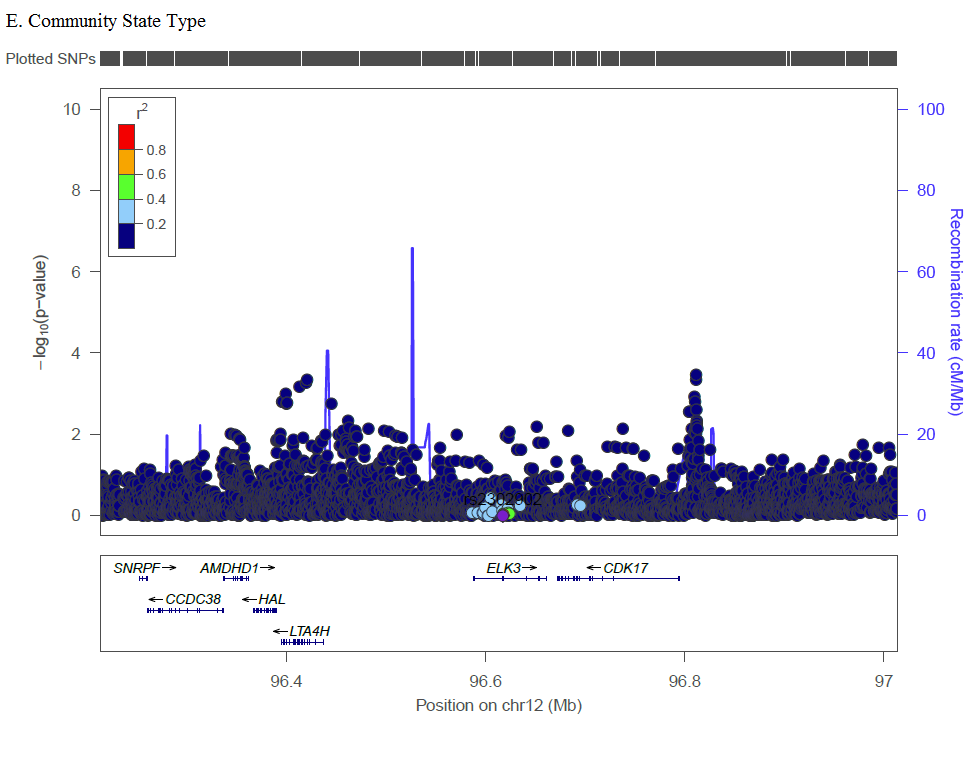
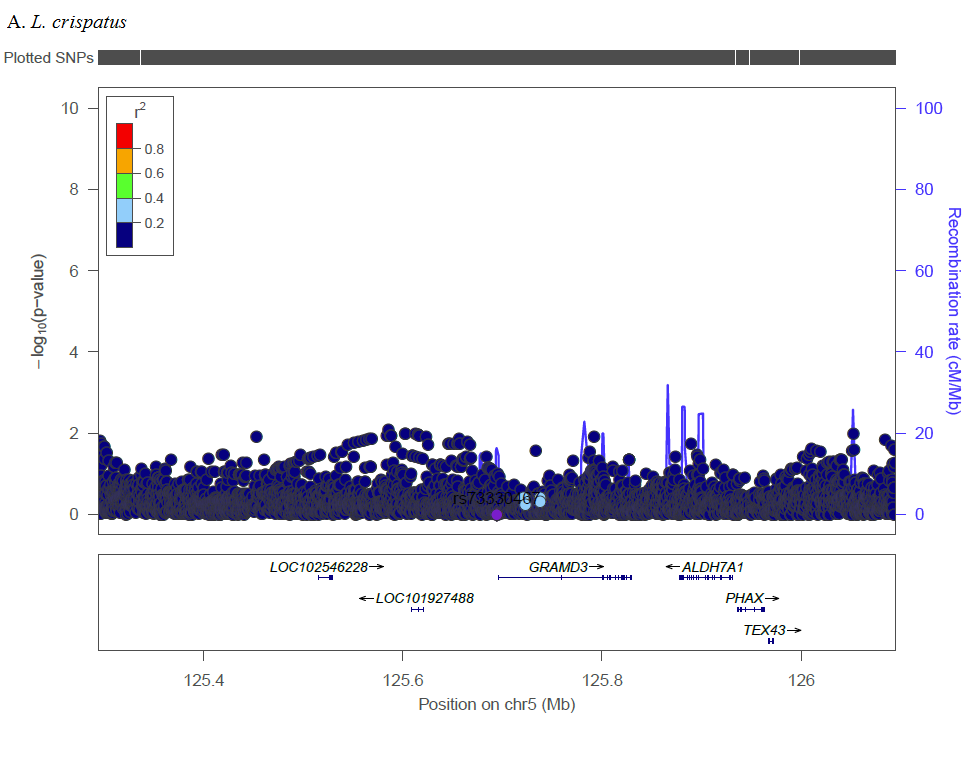
In GWAS, the most significant SNP associated with CST (rs2302902) is located in an intron of ELK3, a member of the ETS transcription factor family, which has not been previously associated with any vaginal microbiome traits or BV. Proteins in the ELK3 subfamily are recruited by serum response factor and participate in transcription regulation, and has been associated in various cancers [19–21], including HPV-positive tumors of oropharyngeal cancer [22]and HPV16 in cervical tumors [23]. In meta-analysis, molecular and clinical measure of BV is associated with two-fold increase in risk of high-grade cervical epithelial neoplasia (CIN) or cancer [24]. Non-optimal vaginal microbiome leads to mucosal disruption and a pro-inflammatory environment that can facilitate HPV acquisition [25], but there may also be underlying shared or synergistic mechanism with ELK3. Other SNPs identified in GWAS occurred primarily in intergenic regions that haven’t previously been associated with BV or vaginal microbiome traits, and should be assessed for replication in future studies.
BV was absent from CST-I (L. crispatus dominant) and infrequent (5.1%) in CST-III. Although CST-IV accounted for 89% of BV cases in this sample, 57% of women in CST-IV did not have BV. This is in keeping with many other studies: women with diverse CSTs are much more likely to have BV, but as many as 50% of women with these CSTs do not have BV [26]. Host genetics may partially explain this variability. For example, women with similar vaginal bacterial colonization (such as those within CST-IV) may have different response to bacterial colonization depending on genetic traits. For women who mount an inflammatory response, this immune response may potentiate BV rather being solely a response to BV. In our study, restricting analyses to HIV uninfected women did not change our findings. This is not entirely unexpected, as associations between HIV and subsequent impact on the vaginal microbiome are variable, with some finding no effect [27, 28]. Conversely, there is substantial data that demonstrate the temporal association between non-optimal vaginal microbiome composition, mucosal inflammation, and subsequent HIV risk [29], and our results suggest that host genetic differences may underpin this. Well-powered longitudinal studies that examine how host genetics is associated with varying CST, mucosal immune markers, and BV trajectories could be informative to understanding this variability in pathology within non-optimal CSTs. Additionally studies are needed to explore the potential long range regulation of these SNPs on genes, as well as evaluate for expression or methylation quantitative trait loci associations.
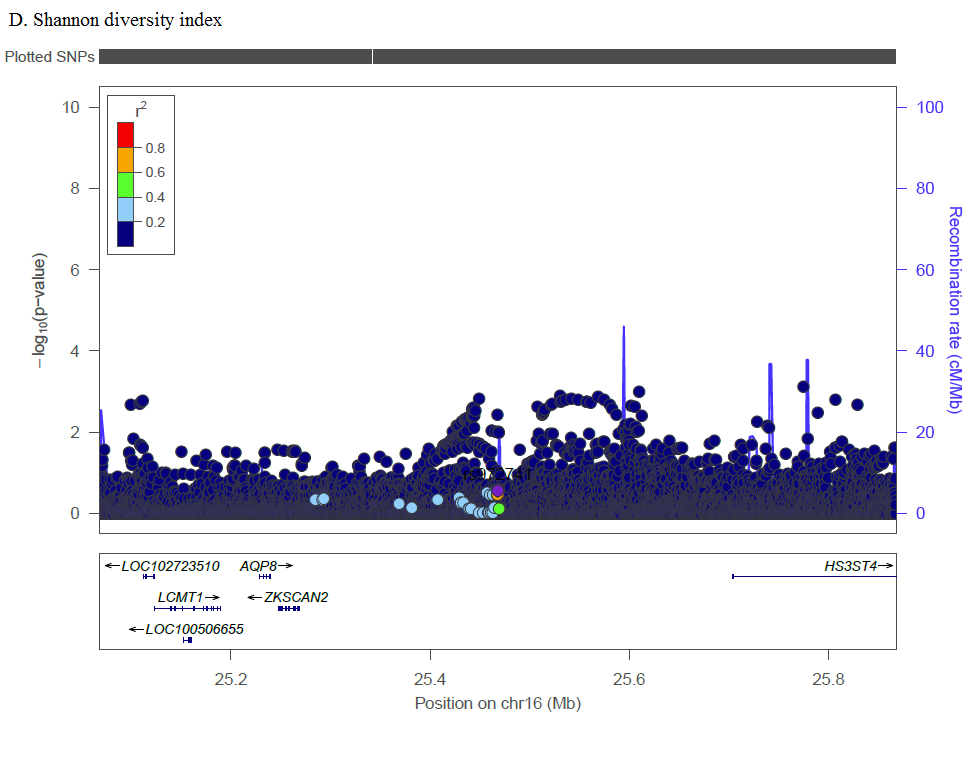
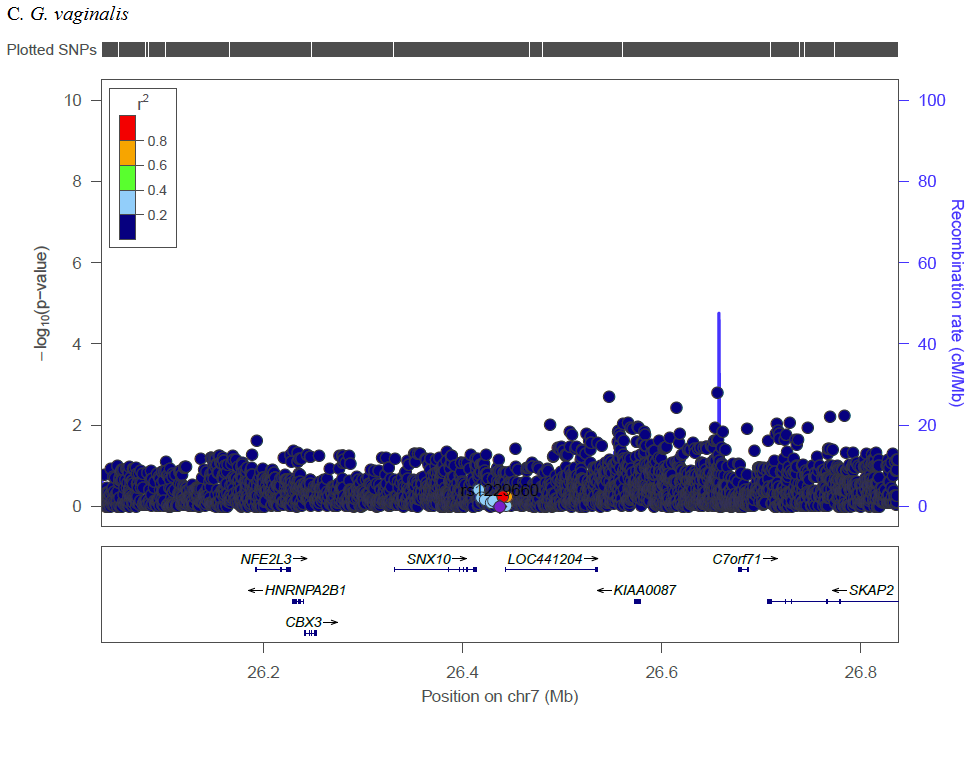
Complex traits are polygenic, and our analysis of previously reported loci identified multiple SNPs across numerous genetic loci with Bonferroni corrected significance. Several SNPs in the MBL2 region were significantly associated with L. iners, G. vaginalis, and CST. Kalia et al. studied these previously reported MBL2 SNPs in association with BV in women in India due to associations between mannose binding lectin (MBL) insufficiency and increased susceptibility to infectious diseases [30]. Among Brazilian women, a specific MBL2 polymorphism was associated with increased odds of recurrent BV compared to controls [31], though a study among Italian women that screened for three specific MBL2 SNPs found no differences for women with recurrent BV as compared to controls [32]. The relevance of this gene may be population specific or subject to interaction with other factors such as the prevalence of co-infections (e.g., HSV-2, HIV), sexual exposures (e.g., partner circumcision status, multiple sex partners, condom use), or non-sexual factors (e.g., cigarette smoking, intravaginal practices). Additionally, we identified SNPs in IL-1RN (CST), IL-5 (Shannon), and IL5RA (L. iners, Shannon). IL-1RN (the gene for interleukin 1 receptor agonist, IL-1RA) inhibits the inflammatory IL-1 cytokines, IL-1α, and IL-1β. In a study of 62 Italian women, compared to women with Nugent score 0–3, women with BV (Nugent score 7–10) had increased IL-1RA, and increased inflammatory IL-5 was associated with decreased Lactobacillus [33]. In candidate gene analysis, Si et al. targeted genetic variants of IL-5 finding association with increased abundances of various Prevotella [34]. Our findings further these studies by analyzing genetic factors and vaginal microbiome traits in a novel population and by demonstrating trans-ethnic associations for these previously identified loci.
In pathway analysis, GPCR pathways were identified with L. crispatus, G. vaginalis, and CST, and Class B/2 (secretin family receptors) within the GPCR family were identified with Shannon diversity index and CST. GPCRs play a significant role in intracellular signaling in response to a broad range of stimuli (e.g., hormones, neurotransmitters, proteins, etc.) and across a wide range of functions (e.g., growth, nutrition requirements, response to disease, etc.) [35]. This likely includes vaginal microbiome modulation, and variation in GPCR signaling may related to variations in innate immune response. LL-37 is elevated in women with BV and in an ex vivo endocervical model, after application of a GPCR inhibitor, LL-37 mediated induction of IL-8 production was inhibited [36]; this is relevant given that IL-8 is often elevated in women with BV [37]. In analyses of previously reported SNPs, we found SNPs in the IL-8 gene region were associated with Shannon diversity index and CST. In an experimental study, Mares et al. demonstrate that application of CVL from women with BV induced higher levels of IL-8 and NFkappaB in human monocytes than CVL from women without BV [38]. Among women in the CAPRISA 004 trial, those with vaginal CST-IV had increased mucosal inflammation (including elevated IL-8) and increased risk of HIV seroconversion [29]. The effects of variation in GPCR signaling on innate immune response may extend to other outcomes. Among Dutch women, GWAS and pathway analysis found that genes encoding GPCR signaling were enriched for 71 Chlamydia trachomatis seropositive women compared to 169 control women [39]. GPCR signaling encompasses a broad range of actions, but in conjunction with our pathway results implicating IL-8 and TLRs, and other studies showing potential roles for GPCR in cervicovaginal health, GPCR signaling merits further study as a potential pathway affecting acquisition or maintenance of vaginal L. crispatus, CST membership, and subsequent BV status.
Our pathway analysis additionally linked neutrophil degranulation with L. crispatus and G. vaginalis. Neutrophils are effector cells in innate immunity and degranulation can have serious adverse consequences for the host due to release of damaging molecules. We are unaware of other studies linking neutrophil degranulation to the vaginal microbiome, BV, or other vaginal infections. However, myeloperoxidase (MPO) is a measure of neutrophils and is elevated in women with sexually transmitted infection (STI) [40]. Whether neutrophil degranulation is an important process in vaginal microbiome composition may be evaluated in subsequent studies, such as by targeting genes encoding molecules associated with this pathway (e.g., β-arrestins, Rac2) [41].
Lastly, pathway analysis identified ‘MyD88:MAL (TIRAP) cascade’ with L. iners, and we identified SNPs within the MyD88 gene in relation to L. iners and Shannon diversity index. MyD88:Mal (TIRAP) is an adaptor to MyD88, the first downstream component of TLR4 and TLR2, and part of the larger overall process for inflammatory cytokine effects [42]. In pathway analysis, several TLR signaling associations were associated with L. iners relative abundance, and in analysis of previously reported SNPs, TLR2, and TLR3 had associations with Shannon diversity index, CST, and L. iners. As summarized in Supplementary Table 1, several investigators have found polymorphisms in TLR genes related to altered risk of BV and enrichment of BV-related bacteria. TLRs, especially TLR2 alone or in combination with TLR-1/TLR-6 and TLR-4, have been recognized for their association with BV and BV-related bacteria, as these TLRs precipitate release of pro-inflammatory cytokines and recruitment of inflammatory cells [43–47]. As summarized by Taylor et al., TLR gene variants can lead to inadequate or excess inflammatory immune response, which can affect disease susceptibility or progression [44]. Additionally studies are needed to evaluate the potential biological role TLRs have on vaginal microbiome composition. Given that the microbiome is a complex trait and complex traits are pleiotropic, this may provide understanding of how TLR variants contribute to risk of BV, response to BV treatment, or other outcomes of non-optimal vaginal microbiome composition, such as adverse pregnancy outcomes. Taken together, our findings demonstrate numerous biologically relevant pathways and phenotypes associated with vaginal microbiome traits.
Limitations
We found substantial functional overlap in several biologically relevant innate immunity pathways. This may in part be driven by the compositionality of individual taxa. To mitigate this, we limited examination to a few traits of known importance in conjunction with comprehensive community characterization. We also confirmed numerous SNPs in close proximity to genomic loci associated with innate immunity reported in previous studies of host genetic traits associated with BV and vaginal taxa. Although we identified novel candidate regions associated with vaginal microbiome traits, additional studies are needed to replicate our findings. Evaluation of functional impact with multi-omic studies (such as gene expression, epigenetics, and fine mapping) would allow mechanistic inference and aid in elucidating regulatory elements, which could lead to translational advances in optimizing the vaginal microbiome and preventing BV and associated consequences.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}