Study Populations
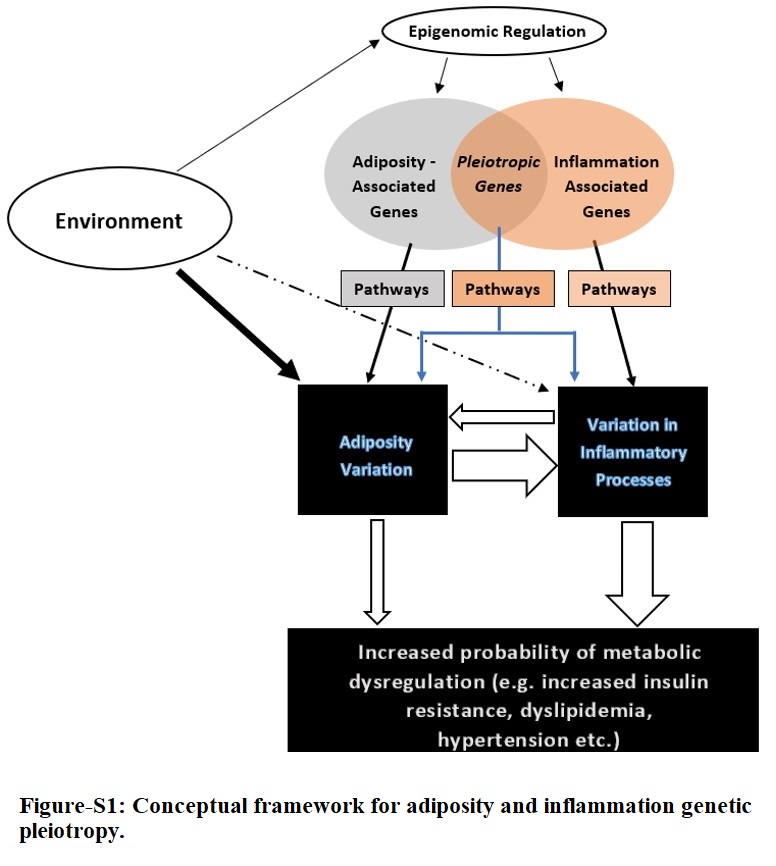
The conceptual framework for the adiposity-inflammation pleiotropy study is displayed in Fig-S1. We chose a discovery and replication study design of self-identified Hispanics study participants from the two studies. The PAGE study population includes several studies at different sites [29, 30] as described previously [31]. In brief, PAGE consists of multiple populations grouped by self-identified ethnicities: European Americans (Non-Hispanic whites [NHW]), African Americans (AA), Hispanics, American Indians (AI), East Asians (ASN), and Native Hawaiians/Pacific Islander (HAW). All participating sites in PAGE ascertained both men and women except for the women only Women’s Health Initiative (WHI). We studied the Hispanics sub-cohort as our discovery set (N = 35,871) from four studies within the PAGE: Hispanic Community Health Study/ Study of Latinos (HCHS/SOL), the Women’s Health Initiative (WHI), BioMe Biobank (BioMe), and MultiEthnic Cohort (MEC). All study participants provided written informed consent and each study was approved by relevant institutional review board. The replication study included self-identified Hispanics study participants recruited at the US-Mexico border, the Cameron County Hispanic Cohort (CCHC). This cohort was established in 2004 and now numbers 5,000 individuals randomly selected from a population with severe health disparities. Many of the participants are originally from Mexico, and we have collected detailed information on participant place of birth, how recently they arrived in the US, income, marital status, and employment [32]. Out of 5,000 individuals recruited for the cohort, a sub sample of 3,313 genotyped individuals are included in this study.
Phenotype measurements and quality control
We studied two anthropometric phenotypes: BMI (kg/m2; a measure of overall adiposity) and WHR (proxy measure of central obesity). For all studies, except in MEC, height and weight were measured by study staff at study enrollment, to calculate BMI (weight/height2). In MEC, BMI is based on self-reported height and weight at enrollment. Pilot analyses of BMI in MEC illustrated a comparable distribution to national surveys [33]. Waist circumference was measured at the level of natural waist in horizontal plane to the nearest 0.5 cm [34]; no waist or hip measurement was available for BioMe sub-cohort; WHR measures were multiplied to 100 for ease of interpretation. Analyses of WHR measures were stratified by sex to account for well-established sexual dimorphism. For inflammation, we used high sensitivity CRP (hsCRP) as robust marker of systemic inflammation with strong association with central obesity [35]; CRP was measured at enrollment separately in each contributing cohort and subsequently harmonized for the entire set [36].
Genotype and quality control
Most individuals (~ 20,000) were genotyped using the MEGA array panel; details on the genotyping were published previously [37]. The remaining PAGE samples were genotyped with Affymetrix and Illumina arrays. We used the genome-wide variants’ set that was imputed to 1000 Genome Phase 3 reference population after quality control (QC), and details are accessible [38]. For this study analyses, we next excluded genetic variants with poor imputation (R2 < 0.4), effective sample size of < 30, and minor allele frequency (MAF) of < 0.05; criteria used for calculating effective sample size for each single nucleotide polymorphisms (SNP) is defined in appendices. Out of > 60 million SNPs, approximately 32 million SNPs were removed because of low MAF. The replication cohort from the CCHC was genotyped at the Vanderbilt University Medical Center genotyping core facility, VANTAGE, using MEGA-EX Array panel. After standard QC, imputation was completed using the Trans-Omics for Precision Medicine (TOPMed) [39] freeze 8 panel available on the NHLBI Imputation Server (https://imputation.biodatacatalyst.nhlbi.nih.gov), by having American ancestry as reference group. We used same QC criteria to exclude SNPs in this replication cohort as substudies from PAGE.
Statistical Analyses
Association tests
The skewed distribution of CRP necessitated log-transformation before regression analyses. Extreme observations (defined as values outside three standard deviations (SD) from the mean in the log transformed distributions (CRP), or 4SD for BMI and WHR) were flagged as outliers and excluded from all analyses. WHR for women and men were treated as separate phenotypes, so that the phenotypes included were CRP, BMI, WHR-men, and WHR-women. Residuals for each phenotype were estimated from linear regression models adjusted for age, age2, sex (when applicable), BMI (when applicable), center, cohort, and age-by-sex interaction, performed separately in each study. Residuals were then inverse rank normalized and used as the outcome for univariate genome-wide association tests (GWAS). All MEGA-genotyped samples were pooled together for genetic association testing while the remaining non-MEGA data were analyzed by study. GWAS were performed using SUGEN, adjusted for 10 genetic principal components (PC). SUGEN employs generalized estimating equations (GEE) to adjust for family relationships (first or second degree), with independent error distributions by self-identified group [40]. GWAS results from MEGA samples and other studies were then meta-analyzed with METAL [41] assuming fixed-effect inverse-variance weighting.
Multi-trait association test
To identify loci with evidence for associations with one or more of the adiposity and inflammation traits, we combined Z-score statistics from meta-analysis results from each phenotype using adaptive Sum of Powered Score (aSPU) [42] test. Briefly, this method aggregates information across n phenotypes for a given SNP by taking the sum of its univariate GWAS Z-scores, each raised to some power γ. As such, a higher γ increases the influence of strongly associated phenotypes on the sum. By allowing γ to take one of many candidate values (1,2…,8), aSPU selects a maximally efficient scheme to detect associations between each SNP and one or more phenotypes.
A set containing trait-specific GWAS-inferred Z-scores (from all contributing phenotypes) was constructed. Multi-trait analysis was performed using the JaSPU (github.com/kaskarn/JaSPU) package, with the number of iterations set to 100 billion. This program relies on a Markov Chain Monte-Carlo (MCMC) [43] iterative process to estimate p-values. The p-values generated from this test were used to assess statistical significance of whether SNPs were associated with one or more phenotypes. The method was selected because it was adaptive to difference in sample size between traits, preserved type 1 error in scenarios, where some other methods did not [42], accommodated direction of effect and computationally efficient which enabled examination of ~ 16 million variants in a reasonable time
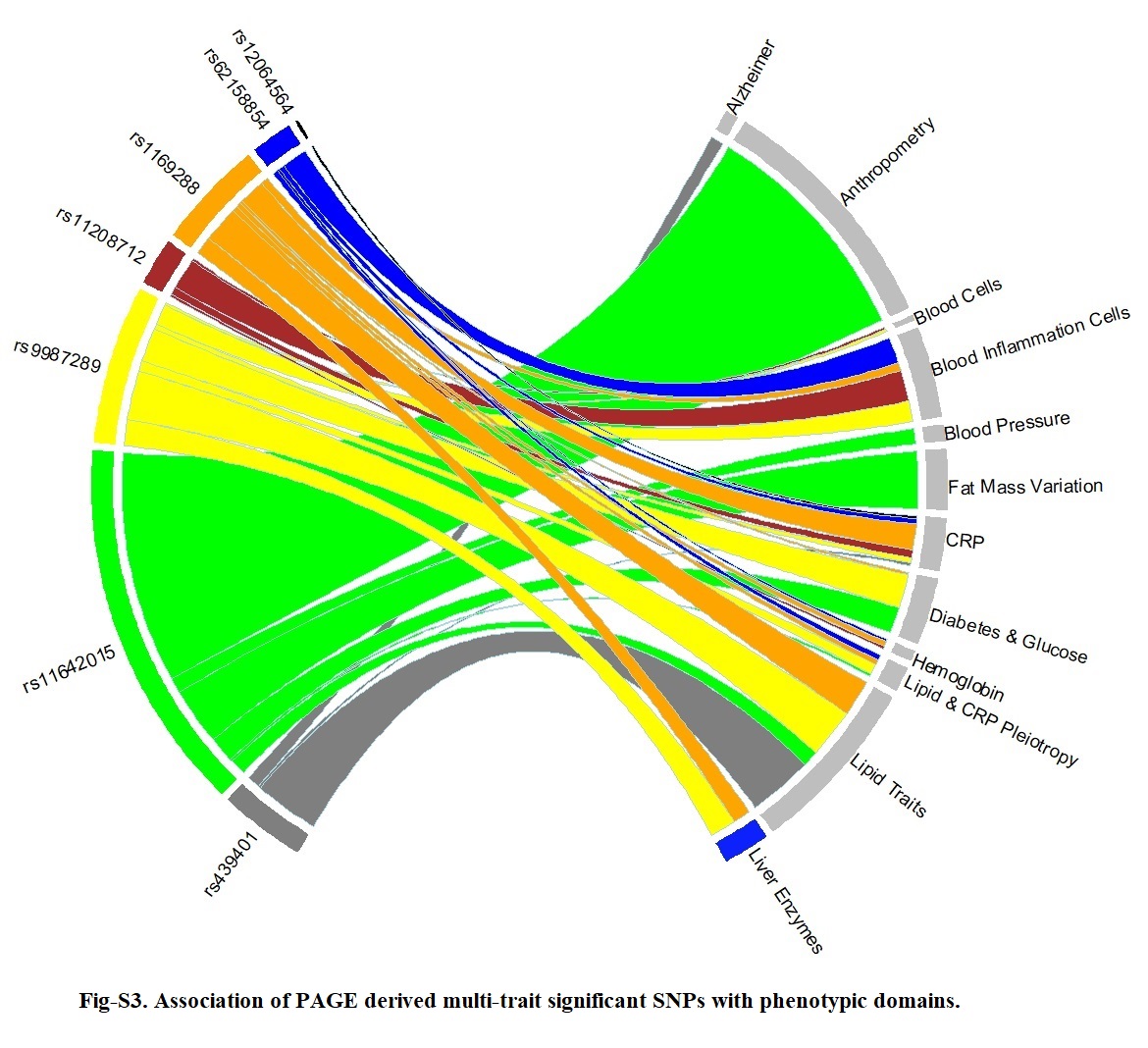
We considered a SNP as a candidate for pleiotropy analyses if a): exceeded significance threshold for multi-trait test at PaSPU < 1.25×10 − 8, a conservative threshold [44] which accounted for multiple testing on likely low frequency SNPs and the number of traits tested, and b) nominally significant for inflammation and at least one adiposity trait in univariate GWAS results. We subsequently identified significantly associated loci where: i) variants in the region met the criteria above, and (ii) were present in pooled MEGA results; latter criterion was used to ensure understudied SNPs existed in both PAGE and CCHC results, thereby usable for likewise comparison. Linkage disequilibrium (LD) analyses identified independent variants (R2 < 0.1). For each independent locus, we highlighted the most likely functional or closest observed proxy to functional variant based on a literature review, and the lowest or close to the lowest PaSPU.
Bioinformatic annotation
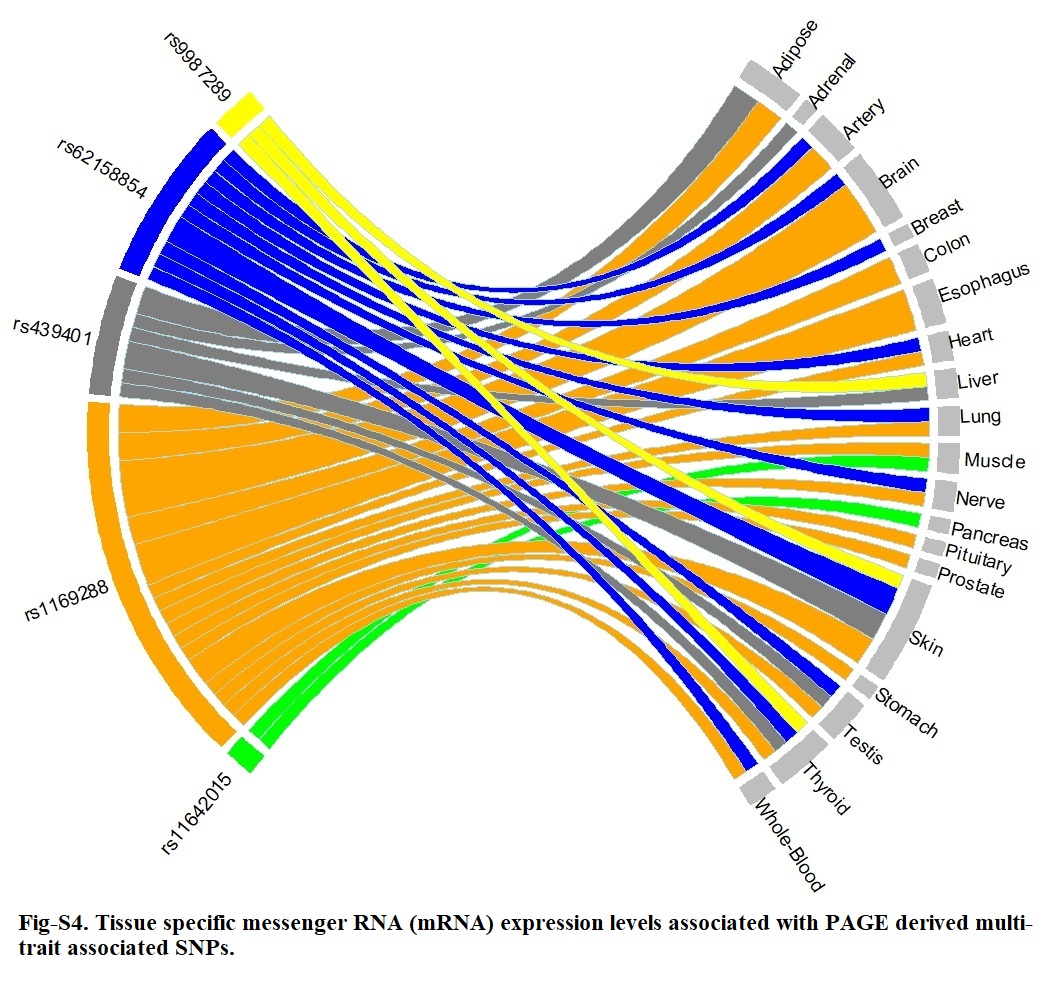
Ensemble Variant Effect Predictor (VEP) (grch37.ensembl.org/) was used to determine the effect of each variant on genes, transcripts, protein sequences, and regulatory regions. We searched replicated variants and their close LD proxies (with LD cut-point of R2 > 0.8) in the publicly available database PhenoScanner [45] and GWAS Catalogue [46] for reported associations with any phenotype including adiposity and inflammation traits at P < 1×10 − 5 significance level. LD clumping was performed using the AMR ancestry panel. We used both Gene Expression Portal (GTEx Portal) [47] and PhenoScanner to examine variants and close proxies (R2 > 0.5) for functional significance and influence on tissue expression at GWAS significance level, which are genomic regions associated with expression levels of messenger RNA (mRNA). Finally, Haploreg was used to evaluate the regulatory potentials of variants on haplotype blocks, such as candidate regulatory SNPs at disease-associated loci [48]. This annotation tool was used to assess the effects of SNPs on regulatory motifs and expression Quantitative Trait Loci (eQTL).
Replication of shared genetic loci
All significant variants were taken forward to replication in the CCHC. GWASs were performed with SUGEN. Multi-trait analysis was completed following the same aSPU based approach used in the discovery stage. The criteria set for replication included, a) variant is nominally significant in multi-trait test (Paspu < 0.05), b) variant was nominally significant for CRP and at least one anthropometric trait, and c) direction of effect of each SNP was same in discovery vs replication cohort for the tested trait.
Causal pathway analyses
We performed a causal mediation pathway analyses [49] of replicated variants to assess pleiotropic potential of identified variants, and whether an observed association is independently associated with several phenotypes and the genetic effect is transmitted through a common pathway that is upstream to the associated phenotypes [50] (i.e., biologic pleiotropy, Fig-S1); or induced due to relationships between the outcome phenotype and a “mediating” phenotype [50]. Elucidating these complexities could provide novel biological insights [51].
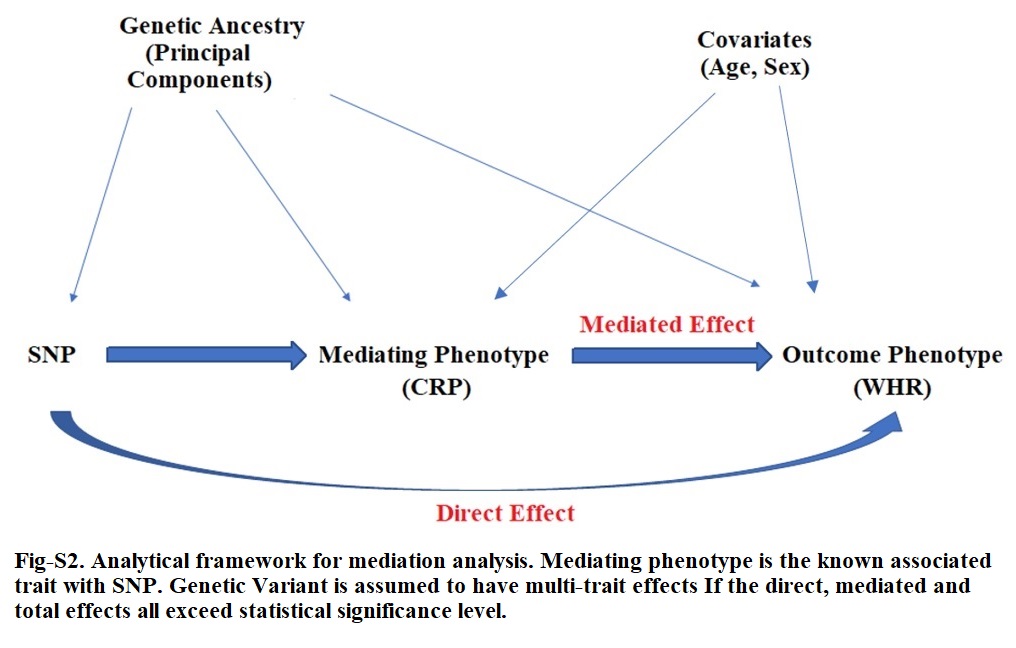
Mediation analyses allows for the assessment of whether a SNP has a direct effect on the phenotype of interest. The R package Mediation [52] was used for analysis, which conducts a three-step process. The total effect between a SNP and an outcome is assumed to be the sum of the average causal mediation effects (ACMEs) and the average causal direct effect (ADE). The first step includes estimating the distribution of the mediator phenotype as a function of the SNP, adjusted for genetic ancestry (e.g., mediator ~ genotype (variant) + PCs + covariates). The next step involves estimating the distribution of the outcome phenotype as a function of the mediator phenotype, genotype, and covariates (e.g., outcome ~ mediator + genotype + PCs + covariates). The final step combines the fitted models in the mediation equation, providing estimates and p-values for the direct (e.g., genotype → adiposity [outcome]) and indirect/mediating (e.g., genotype → mediator → adiposity [outcome]) associations. We did not allow for any mediator by genotype interaction [15]. We performed mediation analyses in the biggest subset of the discovery cohort that were genotyped by the same MEGA array, excluding samples with missingness since mediation method required complete observation across output and mediating phenotypes; the total available sample size stood at 15600. We considered age and sex as covariates in the mediator and outcome models, together with first 10 PCs. We additionally performed sex stratified analyses but 95% confidence intervals and point estimates overlapped with sex adjusted results. With this method, quasi-Bayesian Monte Carlo simulations are used for the estimation of standard errors. The number of simulations was set to 1,000, with robust standard errors using sandwich estimators [53]. We also used same mediation package to further perform sensitivity analysis on the mediated and direct effects for violation of the sequential ignorability assumption and assessing the effect of unmeasured confounders. For genotypes, we used allele dosages and models were estimated assuming additive genetic effects.
For each significantly replicated SNP, if SNP was previously associated with inflammation, we performed mediation analyses assuming CRP as mediator and an adiposity trait as the outcome (Fig-S2); conversely, where a SNP was primarily associated with adiposity in prior studies, then we used adiposity phenotype as the mediator and CRP as the outcome. Statistical significance of ADE in either scenario would suggest an independent association of the lead SNP with the outcome and a potentially biologic pleiotropy effect.
{kind=link}
{kind=link}
{kind=link}
{kind=link}