Phenotypic Characteristics and Cytological Observations of MT and WT
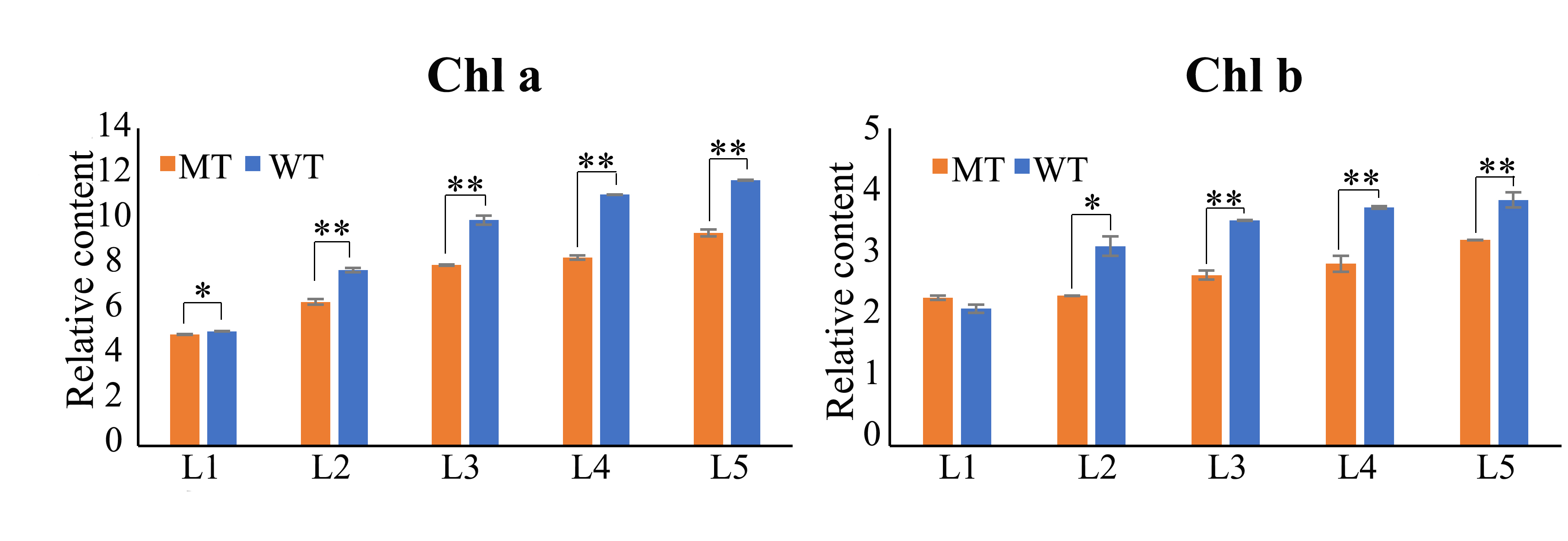
Compared with WT, the PGMS line CCRI9106 (MT) was accompanied by a relatively delay-growth plant and pale green leaf phenotype (Figures 1A). Further, the chlorophyll and carotenoid content were analyzed to determine the pigment difference between MT and WT leaves. The chlorophyll contents of MT leaves were significantly reduced except the chlorophyll b content in the first leaf. Additionally, the second and fourth leaves of MT showed significant reduction of carotenoid (Figures 1B and Supplementary Figures S1). The decrease in pigment content indicates that the pigment synthesis pathway of MT may be defective.
Particularly, the MT flower displayed shorter filaments and shriveled anthers (Figure 1C), with the anthers did not dehisce, and lacked mature pollen grains at the late development stages of anther when stained with 1% I2-KI solution (Figure 1C).
To understand the developmental abnormalities of anther and pollen in MT, light microscopic examination was applied. During the early developmental stages, there were no remarkable morphological differences between the WT and MT. Microspore mother cells (MMCs) underwent meiosis and successfully formed the tetrads of haploid microspores (Figure 1D). Subsequently, at the early uninucleate pollen stage, young microspores were freely released from the tetrads (Figure 1D). With the development of microspores, the WT anther chambers gradually expanded. In contrast, at the same stage, MT had relatively narrow anther chambers (Figure 1D). At binucleate pollen stage, the WT displayed round pollen grains with abundant inclusions, whereas most of MT pollen grains failed to accumulate storage inclusions, and the microspores were irregular in shape (Figure 1D). At the mature pollen stage, the anther endothecium layer of WT began to thicken, preparing for the dehiscence of the anther wall. Consistent with the previous observations, the MT anther wall started to collapse, resulting in unvital pollens. These results showed that the abortive phenotype of MT may be caused by abnormal anther and immature pollens.
Defects of Anther Structure and Pollen Wall Development Cause Male Sterility in MT
Transmission electron microscopy was performed to further observe the ultrastructural changes in MT at various pollen development stages. As revealed via bright-field microscopy, the pollen defects of MT were first observed at uninucleate stage. MT displayed chaotic anther cell layers structure and a thinner pollen nexine when compared with WT (Figure 2C-D, c-d). At binucleate pollen stage, the WT pollen grains had evenly stained cytoplasm which contained numerous small storage bodies (Figure 2E). At higher magnification, the WT nexine was considerable thickened with the tectum and bacula regularly assembled on nexine surface (Figure 2F). In contrast, the MT pollen grains showed severe cytoplasmic degradation (Figure 2e) and abnormal exine assembly with irregular accumulation of tectum and bacula (Figure 2f). At mature pollen stage, these differences were more apparent. Whereas WT pollen grains had accumulated starch granules as well as a mature exine filled with abundant tryphine (Figure 2G, H), MT pollen had a completely empty antrum and absent tryphine (Figure 2g, h). Since it is well known that the synthesis of intine is done under the control of the microspore, it is no wonder that MT aborted pollen had an abnormal intine (Figure 2h). At later stage, the endothecium of WT anther particularly expanded for dehiscence (Figure 2I), yet, MT anther showed a thin endothecium which failed to expand (Figure 2i), resulting in the indehiscence phenotype. Combined with the finds of light microscopy of transverse sections, TSM results confirmed that the male sterility phenotype of MT may be attributed to the irregular exine, immature intine, lacking tryphine and abnormal anther wall development.
RNA-seq for Anthers of Three Developmental Stages based on Cytological Observation
Cytological analysis demonstrated that the abortion of MT pollen occurs during the uninucleate stage (Figure 1,2). Based on this observation, RNA-seq was performed using anther samples in the tetrad (TTP), late uninucleate (LUNP), and binucleate (BNP) to understand the gene expression profile alterations, investigate the underlying mechanisms for the male sterility phenotype. A total of 18 libraries were constructed for the anthers of WT and MT. High throughput sequencing generated more than 50 million clean reads for each sample, which was sufficient for the quantitative analysis of gene expression (Supplementary Table S1). Subsequently, by mapping the clean reads to the upland cotton TM-1 reference genome[41], we obtained exceeded 94% mapped reads in all samples, with up to 85% were unique match reads. Additionally, Q30 ≥ 90.86% and GC contents approximately 43% were found in each sample (Supplementary Table S1). The results above showed that the sequencing data is of high quality.
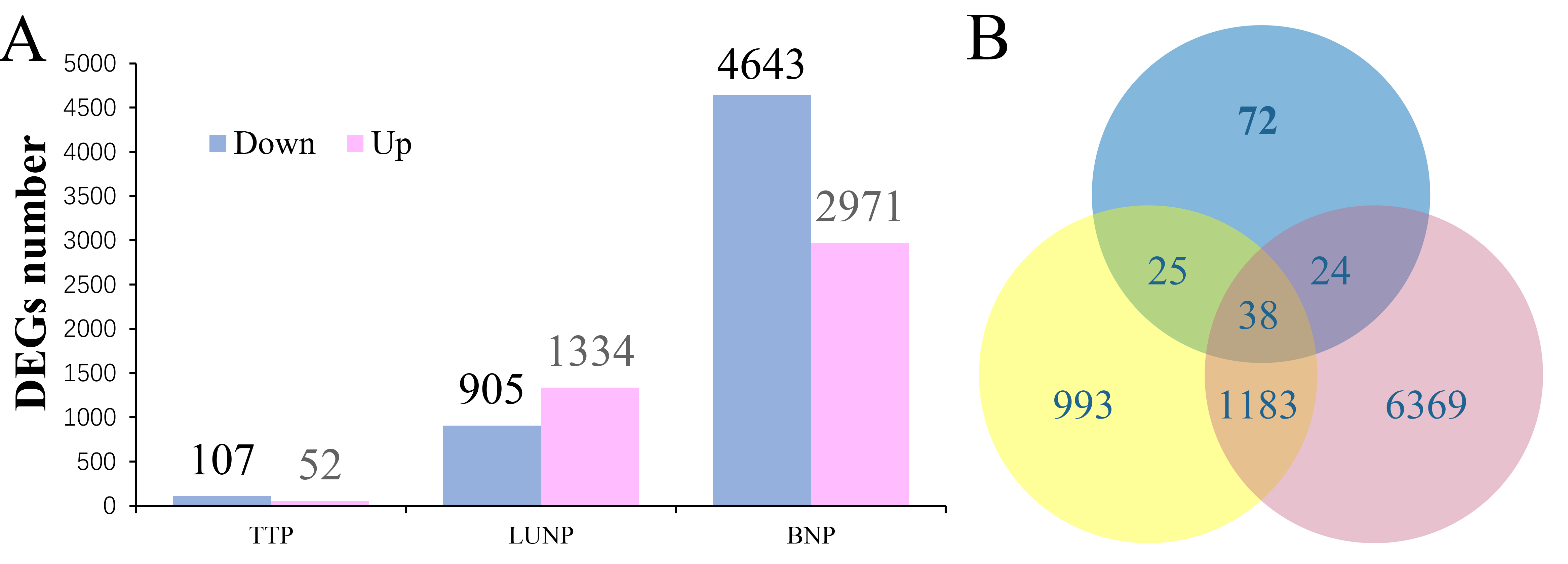
Fragments PerKilo base of transcript per Million mapped reads (FPKM) was used to calculate the gene expression levels. A total of 52,143 unigenes were obtained with the threshold that FPKM>1 in at least one of the samples, of which including 44,993 original genes and 7,150 new genes. Among the original genes, 8,704 differentially expressed genes were filtered with the |log2 fold change|≥1 and FDR≤0.01. 159 (52 up-regulated, 107 down-regulated), 2,239 (1334 up-regulated, 905 down-regulated) and 7,614 (2,971 up-regulated, 4,643 down-regulated) DEGs were found in the MT line at three stages, respectively (Supplementary Figure S2A). The significantly increased number of DEGs from TTP to BNP was consistent with the more serious pollen abortion phenotype observed in the cytological analysis of MT anthers. The number of down-regulated genes were larger than up-regulated genes, indicating that genes related to the anther development were affected. Among these genes, 25 DEGs restricted to TTP and UNP, 1183 to UNP and BNP, 24 to TTP and BNP, and only 38 were differently expression in all three stages, pointing that most DEGs may exhibited specific expression at different stages of anther development (Supplementary Figure S2B).
Down-Regulated DEGs probably Cause the Male Sterility Phenotype of MT based on the Analysis of Gene Expression Patterns
To further provide insights into the functional transitions along anther development, we analyzed the 8,704 DEGs by K-means clustering algorithm, and 11 co-expression clusters were finally obtained. Of these, cluster 3(2561 DEGs, accounting for 29.42% of 8704 genes), in which DEGs specific expressed at BNP of WT, were mostly enriched. The BNP-MT specific expression cluster (cluster 9) was the second enriched cluster with 2,218 DEGs accounting for 25.48% of all DEGs (Figure 3A and Supplementary Table S2). These results indicated that the late pollen maturation process requires precise regulation of large numbers of genes, and the down-regulation of these DEGs may cause the arrest of pollen grains development.
Cluster 1-6 displayed a relatively high transcript level in WT, while DEGs in cluster 7-9 were up-regulated in MT. Cluster 10 was special, for the DEGs showed an earlier expression in MT than WT. DEGs in cluster 11 were particularly enriched at TTP, but exhibited differentially expression at other stages with pretty low levels (Figure 3A and Supplementary Table S2).
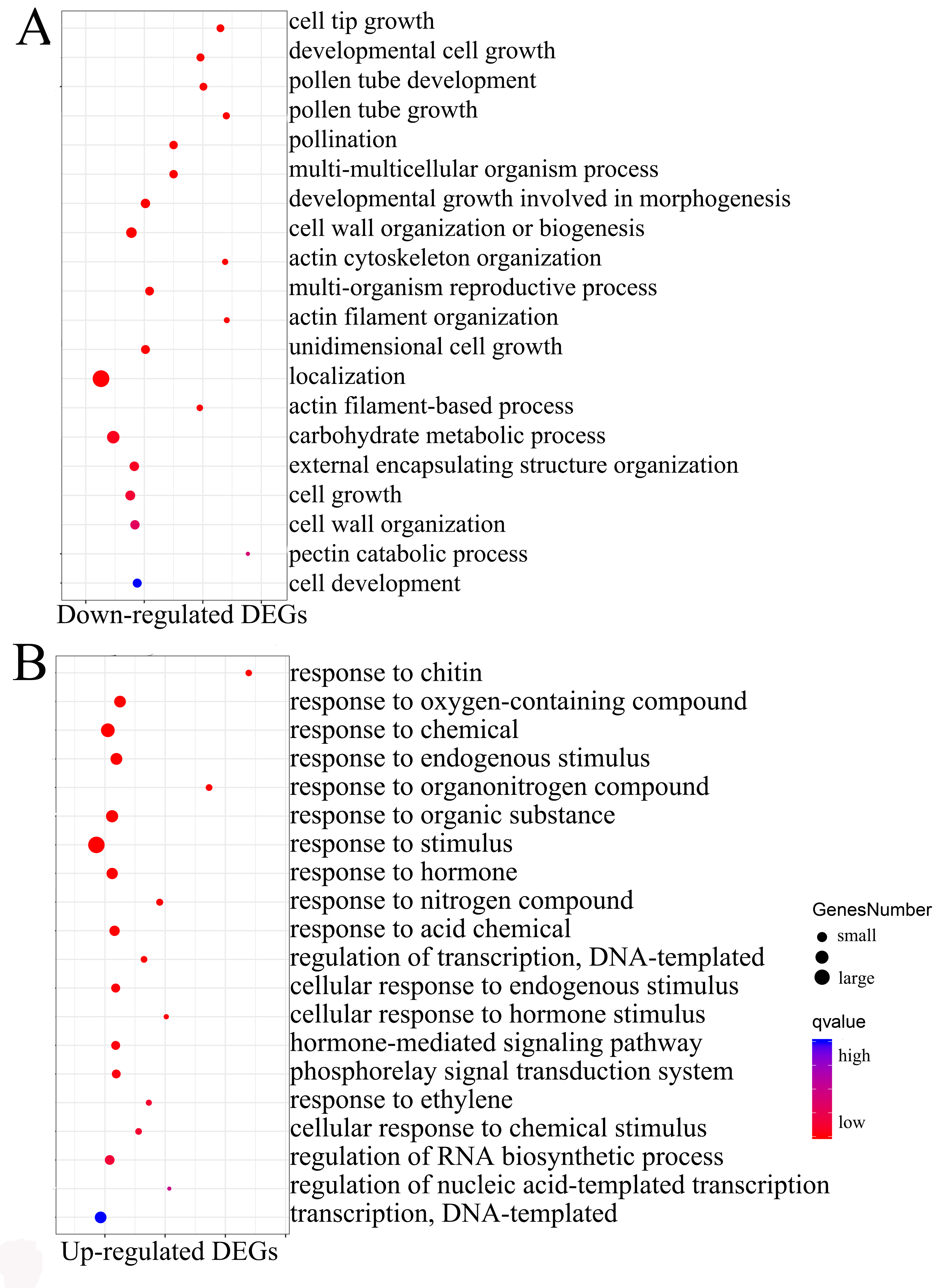
To obtain an overview of the potential functions for DEGs, GO enrichment analysis was performed on all down-regulated (cluster 1-6) and up-regulated (cluster 7-9) DEGs, respectively. The analysis of down-regulated DEGs showed representation of genes related to “cell tip growth”, “pollen tube development”, “cell wall organism process”, “actin cytoskeleton organization” and “carbohydrate metabolic process” (Supplementary Figure S3A). These processes were known to be involved in anther development and pollen maturation. A total of 150 TF-encoding genes belonging to 28 families were found from cluster 1-6 (Supplementary Table S3). The members of MYB, NAC, bHLH, C2H2, LBD and MADS families were highly represented (Figure 3B), and were mainly related to “microsporogenesis”, “floral meristem determinacy”, “pollen maturation” and “secondary cell wall biogenesis” (Figure 3C). In short, enrichment analyses of down-regulated DEGs and TFs indicated that these functional categories are closely related to the male sterile phenotype. Up-regulated DEGs were marked by processes such as “response to stimulus”, “response to hormone”, “response to oxygen-containing compound” and “response to chemical” (Supplementary Figure S3B), in which 394 TFs belonging to 37 families were found (Table S3), and ERF, WRKY, C2H2, MYB, bHLH and NAC families were overrepresented (Figure 3B). GO analysis showed that these TFs mainly response to hormones (ethylene, salicylic acid, jasmonic acid) and stimulus (heat, cold, light or water) (Figure 3C). Taken together, the appearance of these exceptionally GO terms and higher proportions of TFs in cluster 7-9 suggested that the upregulated gene expression may be due to the male sterility phenotype, in other words, the mutant might cause cellular stresses that lead to the general upregulated expression of these genes. Similar founding was reported in previous study[42].
Functional Characteristic of DEGs in Down-Regulated Clusters
Based on the above results, the down-regulated DEGs in cluster 1-6 were more likely to be related to male sterility and were therefore selected for further analysis.
The cluster 1 (95 genes, 0 TFs) corresponds to DEGs that were specifically expressed at TTP and then appeared to be low levels or not expressed at the later stages (Figure 3A and Supplementary Table S2). Previous researches in Arabidopsis and rice supported the idea that pollen exine development, the meiosis of PMC and the degradation of callose walls were processed at the stage around TTP[43]. To explore whether genes in this cluster are beneficial to the above biological process, we performed GO analysis and found the representative significant enrichment terms are “lipid transport”, “external encapsulating structure organization”, “ubiquinone metabolic process”, “carbohydrate metabolic process” and “pollen exine formation” (Figure 3D). These enriched terms suggested that the genes in cluster 1 may be critical for early anther development, which is consistent with previous researches.
Cluster 2 showed peak expression at LUNP, with 565 genes, including 36 TFs (Figure 3A and Supplementary Table S2). GO enrichment showed that DEGs in this cluster mainly associated with “oxidation-reduction”, “secondary metabolic”, “lipid biosynthesis”, “single-organism and terpenoid metabolic process” (Figure 3D). Genes in this cluster may respond to the sterile pollen because the exine and tryphine were assembly with lipidic precursors, and the secondary metabolites and terpenoids are often essential components for tryphine. In addition, nine (25% of 36) MYB TFs (MYB3, MYB7, MYB17, MYB36, MYB48, MYB68, MYB73, MYB85 and MYB105) were found in this cluster (Supplementary Table S2), reflecting their important role in LUNP stage during anther development. Taken together, genes in this cluster may contribute to exine and tryphine formation, and MYB TFs likely take part in these biological progresses as important regulators.
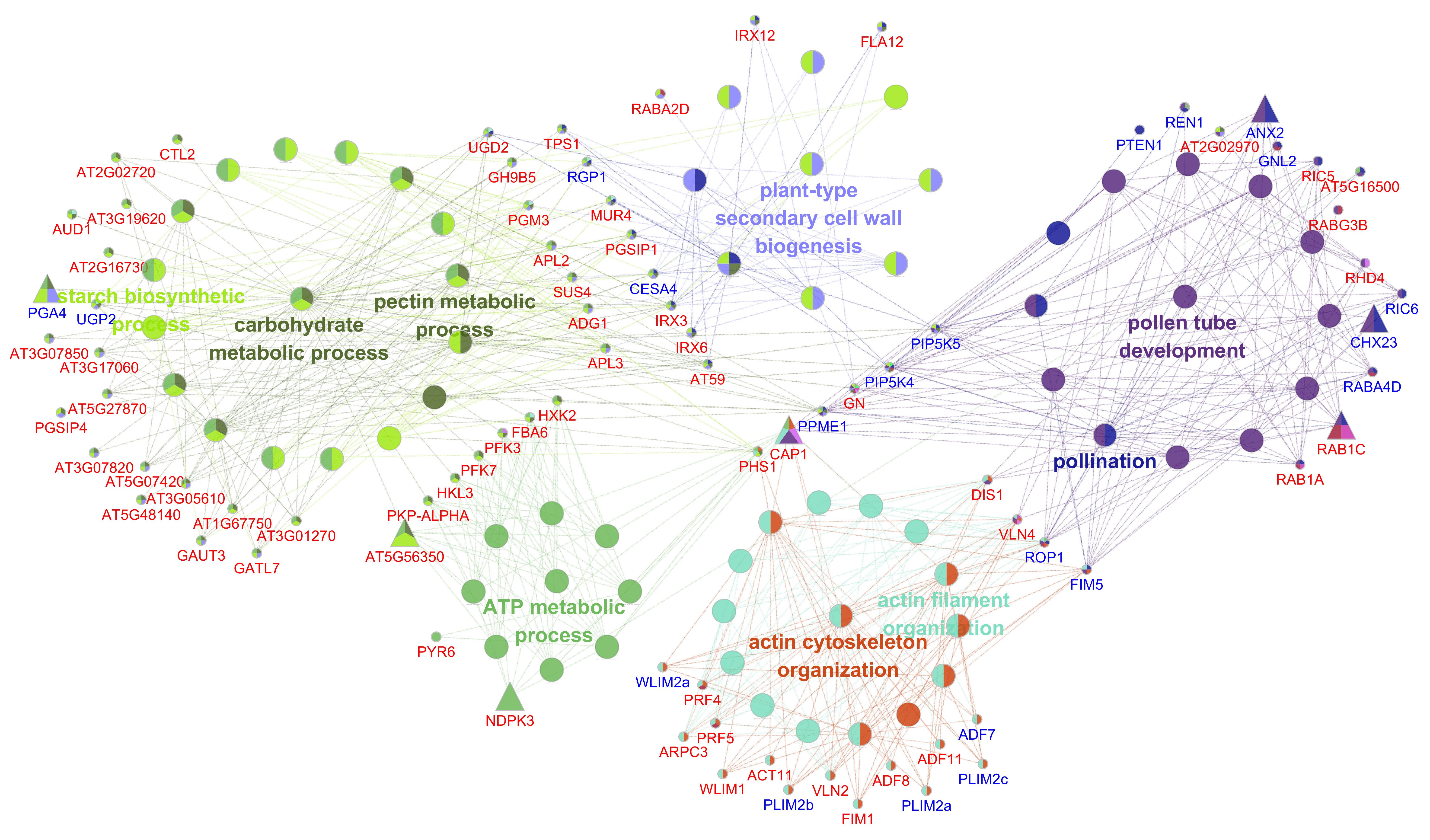
DEGs in cluster 3 were expressed specifically at BNP in WT. 2561 genes, including 68 (2.66% of 2561) TFs were found in this cluster (Figure 3A and Supplementary Table S2), and the appearance of such a large number of DEGs reflected the criticality of this period for anther development. GO terms such as “cell tip growth”, “pollen tube development”, “actin cytoskeleton organization”, “polysaccharide catabolic process”, “pectin catabolic process” and “plant-type secondary cell wall biogenesis” were significantly enriched in this cluster (Figure 3D), indicating that these genes may facilitated the pollen maturation, anther dehiscence and pollen tube growth to some extent.
DEGs in cluster 4 (200 genes, 8 TFs), cluster 5 (160 genes, 4 TFs) and cluster 6 (844 genes, 33 TFs) were expressed at more than one of the three stages (Figure 3A), and all work properly until the down-regulated expression at BNP stage. GO enrichment analysis suggested that these genes may play unique roles for the development of anthers (Figure 3D). These results once again proved that MT anthers at the genetic level showed obvious defects during the BNP stage, and the abnormal expression of these genes during this period may be important cause of male sterility
PPI Network Construction to Identified Candidate Genes in Cluster 1-3
DEG expression pattern clustering and GO enrichment analysis implied that MT male sterility should be controlled by a complex mechanism, and DEGs in clusters 1-3 may be closely related to it. To elucidate this mechanism and further identify key genes and pathways that contribute to MT male sterility, we investigated the known and predicted interactions among genes in cluster 1-3, respectively. Genes in the networks were displayed by their Arabidopsis homologous genes. Functional analysis of those interacting genes was performed using ClueGO software and visualized by Cytoscape 3.3.0.
In network 1, ATA1, AT3G23770, and MEE48 were shown to interact with each other (Supplementary Table S4). ATA1 expressed specifically in tapetal cells and was peaked at the early anther development stage together with AT3G23770[44]. Even though their exact role has not been fully elucidated, the interactions with the proven male sterile gene MEE48 in the network also suggest their probably functions to the anther development.
84 genes were found in network 2 and these genes are related to lipid biosynthetic process and the synthesis pathways of several types of secondary metabolites such as terpenoids, flavonoids and steroids (Figure 4A and Supplementary Table S4). Notably, the productions of these process are exactly precursors for the formation of tryphine, indicating that the formation processes of MT tryphine are indeed abnormal, which is consistent with the cytological defects, thus confirming that the infertility of MT is closely related to genes in this cluster. Additionally, several male sterility genes are present in this network, such as CAS1, ABCD1, KCS6 and LACS1 (Figure 4A). In terms of the abortive phenotype caused by the mutants, we found these genes not only affect the formation of pollen exine and tryphine, but are also required for the formation of anther cuticle[22-25, 45, 46]. Besides, anther cuticle shares similar precursor components with tryphine[9, 10]. Therefore, we considered whether the genes in cluster 2 also response to the formation of anther cuticle, and their down-regulation may affect the anther surface of MT. In order to testify this hypothesis, scanning electron microscope (SEM) analysis was carried out. At pollen maturation stage, the outer surface of the wild-type anther was covered by well-formed cuticle, while the MT anther surface was quite smooth and cuticle seems absent, which is consistent with our inference (Figure 4B). In addition, CHLI, CYP97A3, LUT2, SBPASE, UGT78D2, ABCD1, ECHID and FAB1 were identified as key genes in this network. Of these, CYP97A3 and ABCD1 were reported involved in pollen development[46, 47], indicating that they may be key genes responsible for the male sterility phenotype of MT. Interestingly, several pigment synthesis genes like CHLI1, HEME1, PDS3, CYP97A3, CYP97B3 and LUT2 were found in this network, and involved in multiple biological processes (Figure 4A). The obvious period-specific characteristics in anthers suggesting that they may also play an important role in the development of anthers (Table S4). Further, we used qRT-PCR to detect the expression of several genes in this network, and found the results were similar to those of RNA-seq data (Figure 4C and Supplementary Table S7). These results indicated that genes in this cluster performed various and significant functions on the formation of anther cuticle and tryphine, and their abnormal expression might be the direct cause of the male sterility phenotype of MT.
205 genes were found in network 3, and their involvement are consistent with the GO terms mentioned in cluster 3, confirming their potential roles to the pollen maturation (Supplementary Figure S4 and Supplementary Table S4). Based on the reported MS genes in Arabidopsis[48], several genes which associated with these biological processes were found, such as FIM5, PLIM2a, PLIM2b, PLIM2c, WLIM2, RGP1, UGP2, PGA4, NST1 and CESA4. Numbers of 7 key nodes with high degree were determined and were included in 4 processes (Supplementary Figure S4). Of these, CAP1 may have a central role for the interconnection of these biological processes (Figure S4). CAP1 encodes an actin monomer binding protein that accelerates the exchange of ADP for ATP, is key intermediate between actin-depolymerizing factor (ADF) mediated disassembly and the profilin-based nucleation and elongation machinery, and was reported as essential regulator of pollen tube growth[49]. No TFs were found in this network (Figure S4), indicating that DEGs in this cluster were structural genes located downstream. Down-regulation of these genes may be the results of microspore abortion, in other words, they may be needed for pollen grains maturation and pollen tube growth rather than microspore development.
Hub Genes Identification through WGCNA
Based on the analysis above, we speculated that the down-regulation of genes specifically expressed at LUNP stage are critical to the phenotype of MT, and several key genes associated with virous biological process were identified. But the regulatory mechanism is poor understood. Therefore, weighted correlation networks were constructed for the DEGs based on the pairwise correlations between genes in their common expression trends across all samples to identify key genes and the potential mechanism that are highly associated with the male sterile phenotype of MT, and 10 distinct modules were classified. The module eigengenes for 10 modules were correlated with different samples (Figure 5A).
Of particular interest to us is the LUNP anther specific expression module (MEyellow) (Figure 5B), which contains 526 genes, of these including 31 TFs (Supplementary Table S5). For these genes, 461 (87.6% of 526) were interacting genes in the PPI network of cluster 2 (Figure 4A and Supplementary Table S5), the closely related community formed by these genes indicate their important role to LUNP pollen development. To further detected hub genes of the yellow module, a co-expression network was obtained and then visualized by Cytoscape 3.3.0, and the genes with higher connectivity were showed in Figure 6A. Furthermore, several genes were identified as hub genes. Strikingly, most of them are directly related to lipids. Ghir_D07G012910 (NPC2), a non-specific phospholipase C2 protein with the central location in the network, is involved in gametophyte development. In Arabidopsis, double mutants of npc2-1 npc6-2 exhibited reduced viability of ovules and pollens[50]. A small rubber particle protein3 (Ghir_D11G032630) which associates with lipid droplet surfaces, was reported to be a positive regulator of tissue growth and development and was induced by ABA[51]. Other highly connected hub genes include 2 glycosylphosphatidylinositol-anchored lipid protein transfer 1 ( Ghir_D04G019930/Ghir_A04G015210 ) which bind to lipids and function as a component of the cuticular lipid export machinery that performs extensive export of intracellular lipids from epidermal cells to the surface to build the cuticular wax layer and silique walls[52], a LTP family protein Ghir_A08G018060 with unclear function, and a isoprenoid biosynthesis enzyme DXR (Ghir_D09G017510) which caused a seedling lethal, albino phenotype when were knockout or strongly silenced[53]. Interestingly, Ghir_D13G017030 (CYP97A3) and Ghir_D10G003560 (CHLI1) were also identified as hub genes, which is consistent with the results of the PPI network 2 (Figure 4A, 6A), thus highlighting them as possible key regulators of the developing anther.
In this network, 10 MYB, 4 NAC, 4 LBD, 3 C2H2, 3 bHLH, 2 MIKC_MADS, 2 ERF, 1 GRF, 1 Dof and 1 B3 were found (Supplementary Table S5). Therefore, MYB TFs which with the highest proportion (32.26%) were considered to have an important role in UNP pollen development. In figure 6A, MYB3 (Ghir_A08G003880), MYB7 (Ghir_A07G002060) and MYB48 (Ghir_A13G024180) which included in the PPI network of cluster 2 were also found in this module, indicating these potential roles to the LUNP anther development. In addition, Ghir_A12G017450 (MYB16) was showed a higher connection with hub genes (Figure 6A). Taken together, MYB TFs may be crucial upstream key genes regulating the expression of other genes in this module, and further became the facilitators to the achievement of anther cuticle and pollen tryphine during LUNP stage. Additionally, these hub genes were taken to perform quantitative RT-PCR. The significantly down-regulation of these genes indicated their potential roles to LUNP anther development (Figure 6B and Supplementary Table S7).
ABA Signal Pathway was Altered in MT Anthers
Previous studies demonstrated that ABA plays an important role in the regulation of plant cuticle formation, and ABA biosynthesis and signaling transduction were impaired in mutants with disrupted cuticle biosynthesis[54].
In this study, we found several hub genes were shown to be regulated by ABA, such as Ghir_A01G008890 (AT1G16310), Ghir_D01G009320 (AT1G16310), Ghir_D07G021480 (AT3G25280), Ghir_D08G014410 (MAKR6), Ghir_D11G032630 (AT3G05500), together with Ghir_A07G002060 (MYB7), Ghir_D10G003560 (CHLI1) and Ghir_A12G017450 (MYB16) (Figure 6A). Further, we analyzed the functions of all DEGs in this module based on the homologous annotations in Arabidopsis and found a total of 28 genes in this module have been shown to be involved in the biosynthesis, transport or response of ABA (Supplementary Table S6), of these, Ghir_A07G002060 (MYB7), Ghir_D10G003560 (CHLI1), Ghir_D12G021400 (CYP707A3), Ghir_D13G004890 (FAAH), Ghir_A01G017870 (KCS6) and Ghir_D01G019360 (KCS6) were also found in the PPI network of cluster 2 (Figure 4A), indicating that ABA signal may be a key regulator to LUNP anther development. Meanwhile, the expression trends of several genes assessed by qPCR (Figure 6C and Supplementary Table S7) were consistent with the results of RNA-seq, supporting our deduction. Furthermore, the ABA content in WT and MT anthers was measured during different development stages, and the results suggested that the ABA content of MT anthers decreased across all stages (Figure 6D). These results suggested that ABA signaling process was affected during MT pollen development, and the reduced endogenous ABA levels might lead to the inhibition of genes involved in the formation of anther cuticle, pollen exine and tryphine.
Analysis of LUNP-specific Down-regulated Genes in MT under two Photoperiods
Compared with the male sterile phenotype in Anyang, the PGMS line CCRI9106 is male fertile under short-day conditions when planted in Sanya, China[55]. To examine whether the different phenotypes were reflected at the transcriptional level, the mRNA expression levels of 34 LUNP-specific down-regulated genes, which held higher connectivity in the PPI network and WGCNA module, were analyzed by qRT-PCR. Anther samples of the same stages (termed as TTP-S, LUNP-S and BNP-S) of MT in Sanya were collected for comparison analysis. As shown in Figure 7, most of these genes were up-regulated in Sanya condition, indicating their activation by short photoperiod. HEME1, CYP97B3_A07, KCR1 and AAO3 displayed higher expression levels in Sanya than in Anyang at all stages and shed significant activation in TTP-S stage. While CHLI1, FLS1, MYB7, FAB1 and TT4 exhibited a constantly increasing expression to a peak at the BNP-S stage. DXR, Ghir_D11G032630, UGT78D2_D04, UGT78D2_A05 and LUT2 showed down-regulation in Sanya condition, especially under the last two stages. The remaining genes, which accounted for a large proportion, were specifically up-regulated at LUNP-S stage, even though some of them showed slightly down-regulation during BNP-S stage. Therefore, the differences in the expression levels of these genes under the two conditions indicated that the conversion of fertility was closely related to the functional expression of these genes, and the LUNP stage may be crucial for PGMS line CCRI9106 to respond to photoperiod regulation during pollen development.
{kind=link}
{kind=link}
{kind=link}
{kind=link}