Patient samples
Two cohorts of NPC tissue samples were collected at Hunan Cancer Hospital, Changsha. The first cohort included 38 NPC tissues and 16 chronic nasopharyngeal inflammation tissues (Table S1) which were used for quantitative real-time polymerase chain reaction (qRT-PCR). The second cohort included 99 NPC tissues and 46 adjacent non-NPC tissues (Table S2) for in situ hybridization (ISH). This study was approved by the Joint Ethics Committee of Central South University, and informed consents were obtained from all participants.
Cell culture and cell transfection
Cell lines (HNE2, CNE2, and HONE1) were obtained from the Cell Center of Central South University, and cultured in RPMI-1640 (Gibco, USA) containing 10% fetal bovine serum (Gibco, USA) at 37°C and 5% CO2.
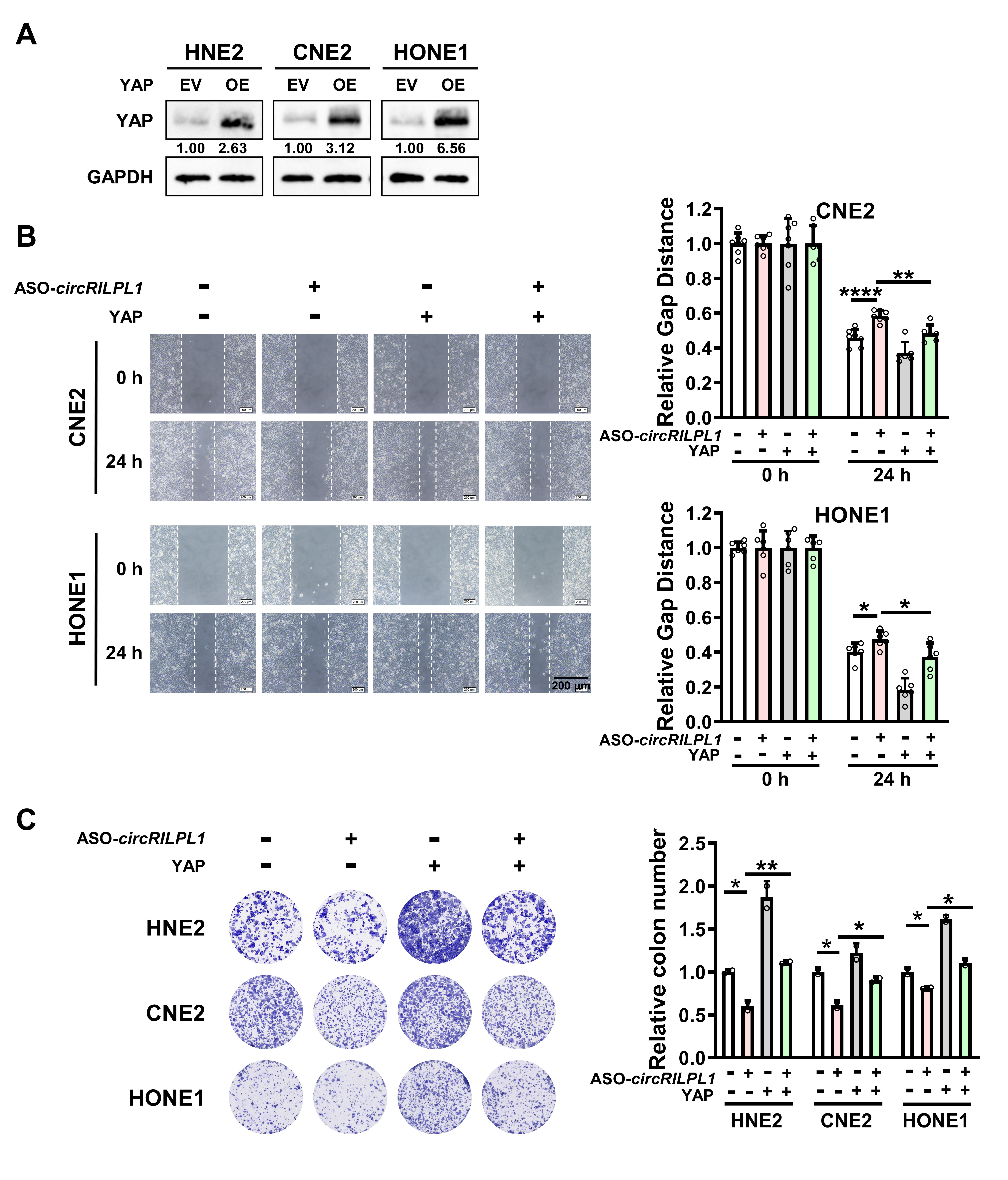
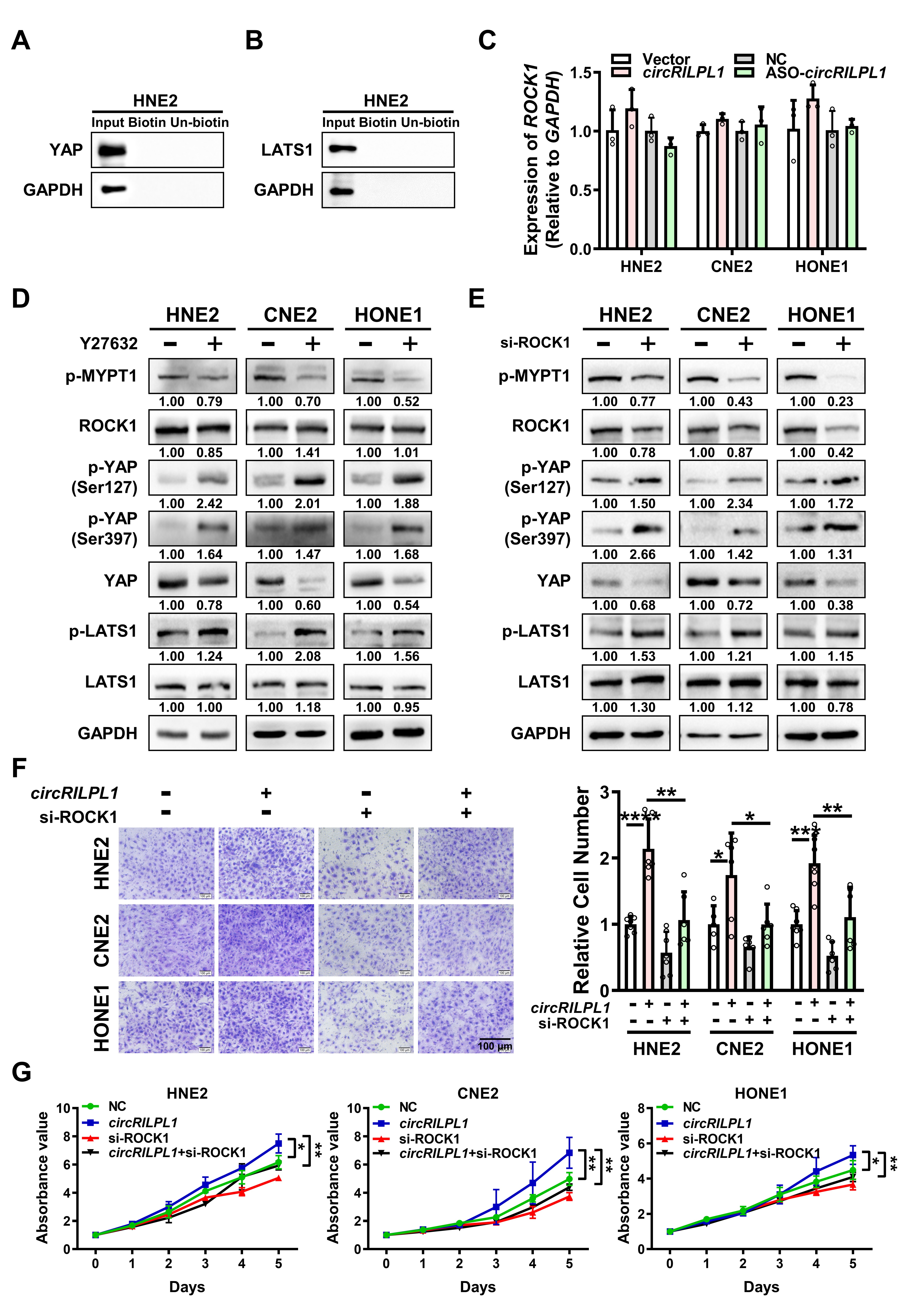
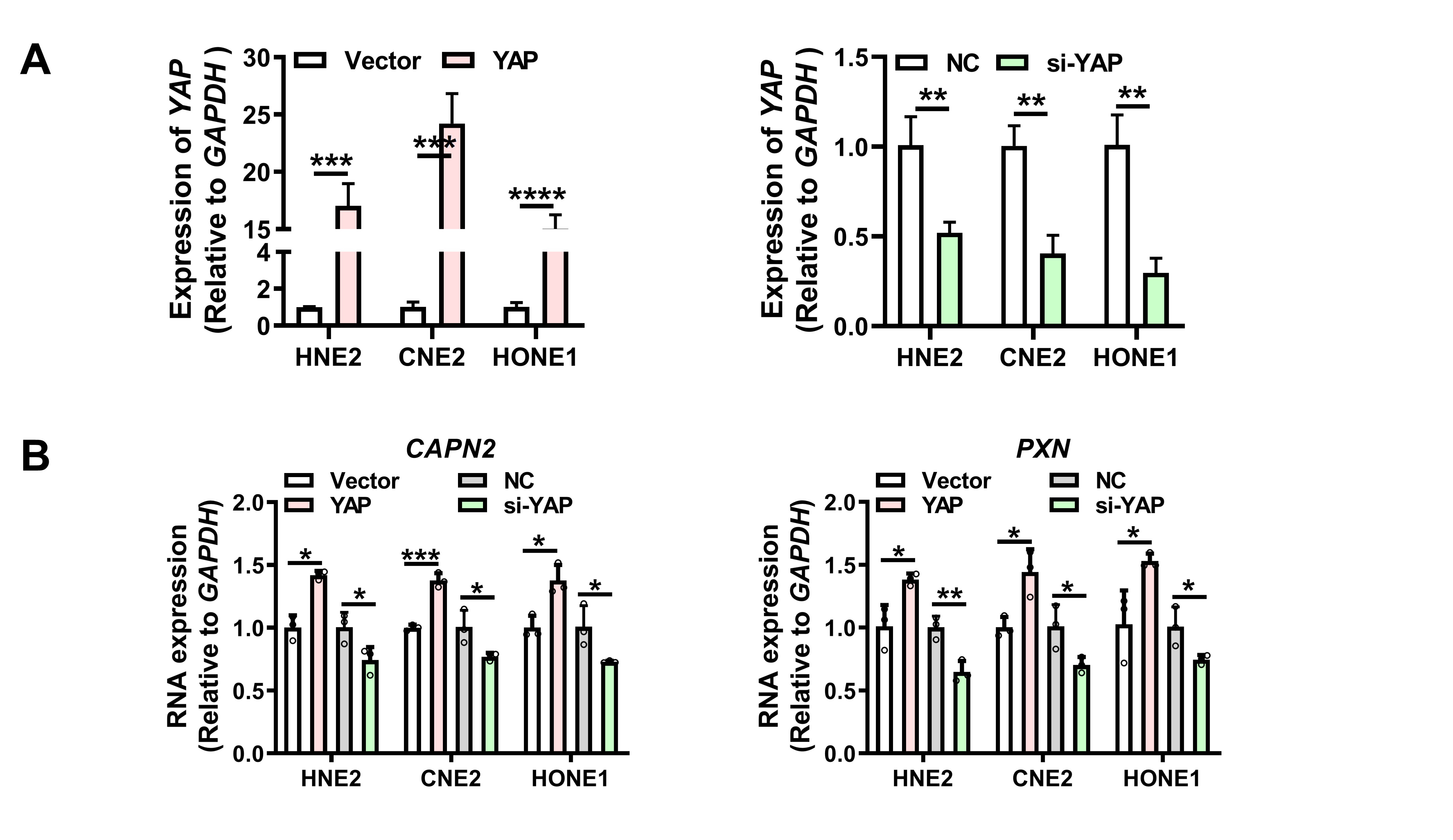
The full-length circRILPL1 (hsa_circ_0007552) was amplified by PCR and then inserted into pcDNA3.1(+) circRNA Mini Vector, which was a gift from Professor Li Yong at Baylor College of Medicine. The YAP cDNA was cloned into pcDNA3.1(+). The construct for overexpression of IPO7 (IPO7 pcDNA3.1-3xFlag-C) was purchased from YouBio Company (Changsha, China). All constructs were confirmed by sequencing. Antisense oligonucleotides (ASO) specifically targeting circRILPL1, siRNAs targeting YAP, IPO7, and ROCK1 [20], and the controls were purchased from RiboBio Co., Ltd. (Guangzhou, China). The sequences for siRNA were listed in Table S3. Neofect (Neofect biotech Co., Ltd. China) was used for plasmid transfection. Hiperfect (Qiagen, Hilden, Germany) was used to transfect ASOs and siRNAs. The ROCK1 inhibitor Y-27632 (Selleck, Shanghai, China) was used in cells with a concentration of 10 uM.
RNA extraction and qRT-PCR
Total RNA was extracted from cells or tissues using TRIzol (Life, USA). Reverse transcription was performed using the HiScript cDNA Synthesis kit (Vazyme, Nanjing, China). Then, qRT-PCR was performed on CFX96™ Real-Time PCR Detection System using 2×SYBR Green qPCR Master Mix (Bimake, USA). All primers used were listed in Table S3.
Western blotting
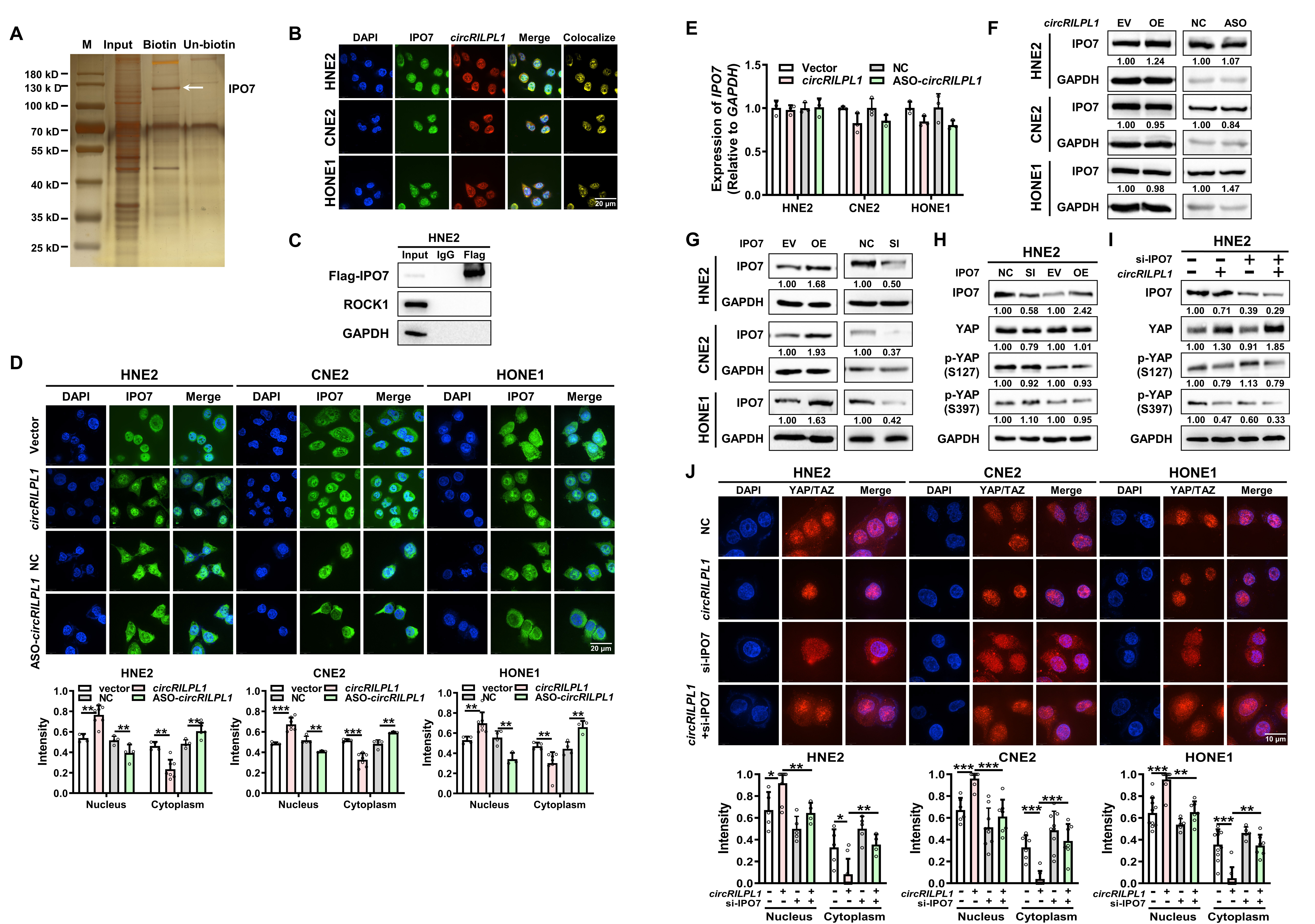
RIPA buffer (Beyotime Biotechnology, Shanghai, China) and Protease Inhibitor cocktail (Roche Applied Sciences, Mannheim, Germany) were used for cell lysis and protein extraction. Protein samples (30-50 μg) were separated via 10-12% SDS-PAGE and transferred onto 0.2 μm PVDF membrane (Millipore, Billerica, MA, USA). After blocking with 5% nonfat milk for 1 h, the membrane was incubated with primary and secondary antibodies sequentially following the manufacturer's instruction. After washing with tris-buffered saline (TBS) or phosphate-buffered saline (PBS) supplemented with 0.1% Tween 20 for 3 times, the target protein bands were detected using the ECL detection system (Millipore, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. All antibodies used are listed in Table S4.
RNase R and actinomycin D treatment
Actinomycin D (Sigma, USA) was added into NPC cells at a final concentration of 2 μg/ml, and RNA was collected at 0 h, 8 h, 16 h, and 24 h for qRT-PCR. For RNase R (RNR07250, Epicentre, USA) digestion, 20 U/μl RNase R was incubated with RNA extracted from NPC cells at 37°C for 30 min, and then the enzyme was inactivated by heating at 70°C for 10 min.
Cytosolic/nuclear fraction assay
Cytosolic and nuclear RNAs were isolated using the PARIS™ Protein and RNA Isolation System (Invitrogen, USA) following to the manufacturer's instructions. Cytosolic and nuclear proteins were separated using the NE-PER Nuclear and Cytoplasmic Extraction Reagent (Thermo Scientific, USA).
Immunohistochemistry (IHC) and in situ hybridization (ISH)
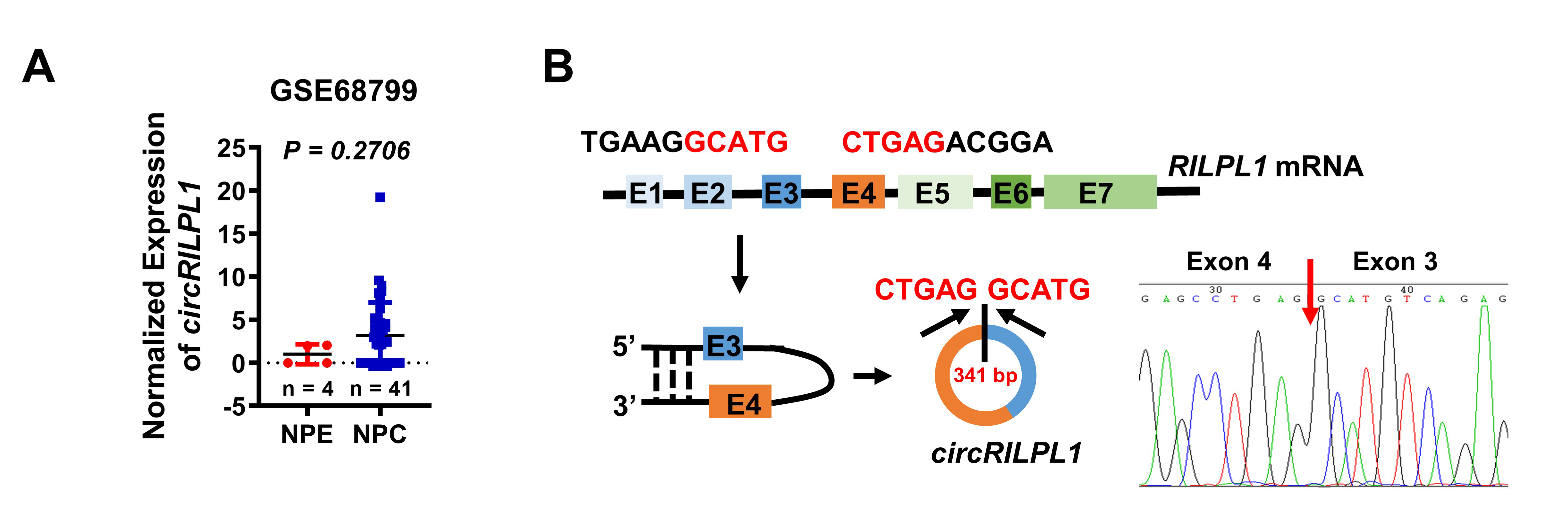
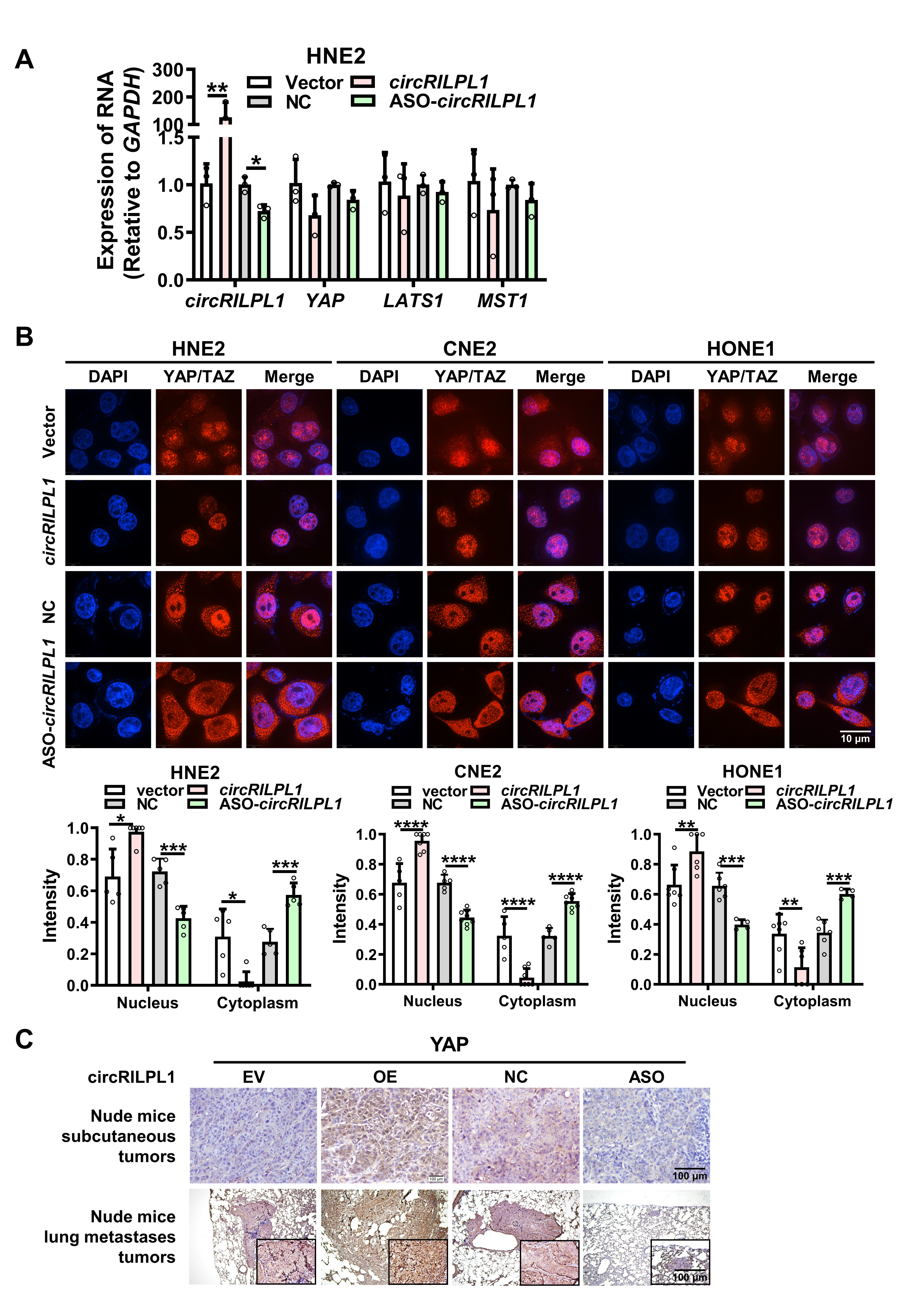
IHC was done using an immunohistochemical kit (KIT-9720, MXB Biotechnologies, Fuzhou, China). For ISH, digoxigenin-labeled circRILPL1-specific probes were synthesized by Sangon Biotech (Shanghai, China) and the expression of circRILPL1 in NPC tissue was detected specimens using the Enhanced Sensitive ISH Detection kit I (POD) (MK1030, BOSTER, China) following the manufacturer's instruction. The probes used were listed in Table S3. All sections were independently scored by two pathologists who were blind to the clinicopathological features of the samples (Table S2). A semi-quantitative scoring criterion was used based on the staining intensity and the proportion of positive cells. When the tissue was not stained, scoring 0. When the tissue was pale yellow, scoring 1; light brown, scoring 2; dark brown, scoring 3. On the other hand, when less than 25% of cells were positive, scoring 0; 25% - 50% positive, scoring 1; 50% - 75% positive, scoring 2; and more than 75% positive, scoring 3. Finally, a comprehensive score was calculated as the product of the staining intensity score and the positive ratio score. Scores greater than 5 were determined as high expression; others were considered as low expression.
RNA fluorescence in situ hybridization (FISH)
Cells were fixed with 4% paraformaldehyde and penetrated with 0.1% Triton-100. After incubation with pre-hybridization solution for 2-4 h at 37°C, cells were incubated with digoxigenin-labeled circRILPL1-specific probes (Sangon Biotech, China) at 37 °C overnight. After washing with SSC buffer, cells were incubated with biotinylated mouse anti-digoxigenin antibody for 1 h. Red fluorescence labeled mouse secondary antibody (LIFE, USA) was added at a dilution ratio of 1:200, and further incubated in the dark at 37°C for 1 h. DAPI (Invitrogen, USA) was added to counterstain the nuclei. Cells were imaged and analyzed using confocal microscope Ultra-View Vox (Perkin-Elmer, USA).
Wound healing and transwell assays
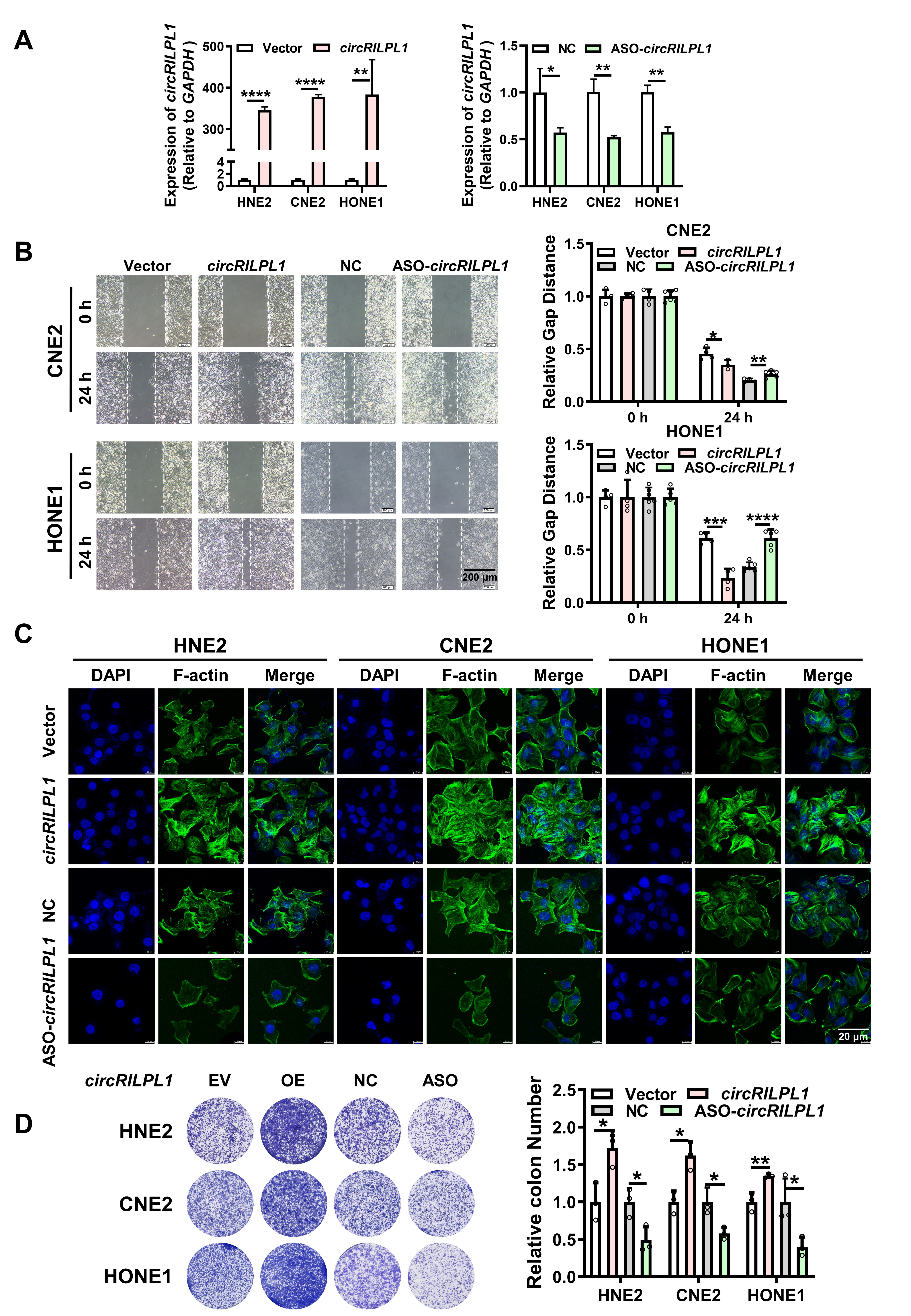
For wound healing assay, transfected cells were plated in 6-well plateand scratched evenly using sterilized mini tips. Then cells in each well were photographed and counted at 0 h and 24 h. For transwell assay, the diluted Matrigel (BD, Shanghai, China) was added to the upper chamber of Transwell chamber (Millipore, USA). Then transfected cells were added to the upper chamber and RPMI-1640 medium containing 20% FBS was added to the bottom chamber. Two days later, cells in the bottom chamber were photographed and counted using an inverted phase-contrast microscope after fixation and staining with 0.1% crystal violet.
MTT and colony formation assays
For MTT assay, 800 transfected cells were seeded in 96-well plates with 5 replicates in each group. After cell adhesion, 20 ul MTT (Beyotime, China) was added and incubated in the dark for 4 h at 37°C. Then, the plate was scanned at 490 nm using a microplate reader from day 0 to day 6. For colony formation assay, 2,000 transfected cells were seeded in 12-well plate and cultured for a week. The colonies in each well were photographed and counted after washing with PBS, fixation with 4% paraformaldehyde, and staining with 0.1% crystal violet.
Atomic Force Microscopy (AFM)
AFM (JPK NanoWizard 4 BioScience, JPK Instruments, Germany) was used to measure the biophysical properties of NPC cells. Cells were fixed in 2% glutaraldehyde for 45 s, 4% paraformaldehyde for 20 min, and then washed and maintained in appropriate amount of PBS for AFM scanning. The probe HYDRA6V-100NG (AppNano, CA, USA) with a spring constant of 0.292 N/m was used in the experiments. The indentation process was performed at a loading and retraction rate of approximately 2.5 μm/s with an indentation depth of at least 1 mm. Images were captured under QI mode, and analyzed with JPK software to obtain data on cell adhesion, stiffness, and Rq.
Immunofluorescence (IF)
Cells were fixed with pre-warmed 4% paraformaldehyde, penetrated with 0.1% Triton-100, and blocked with 5% calf serum albumin for 30 min at room temperature. Then cells were incubated with primary antibodies at 4°C overnight. After washing with PBS, they were incubated with fluorescently labeled secondary antibody (LIFE, USA) at 37°C for 1 h. Images were captured with laser confocal microscope Ultra-View Vox (Perkin-Elmer, USA) after counterstaining the nuclei with DAPI.
Luciferase reporter assay
Cells were co-transfected with the YAP luciferase reporter plasmid and the pRL-TK plasmid (which expresses Renilla luciferase as an internal control) after overexpression or knockdown of circRILPL1. The luciferase activity was measured using the Dual-Luciferase® Reporter Assay System (E1910, Promega, USA) 48 hours later. Relative luciferase activity was obtained by normalizing with the Renilla luciferase activity.
RNA immunoprecipitation (RIP) and chromatin immunoprecipitation (CHIP)
RIP was performed to analyze the interaction between circRILPL1 and ROCK1 or IPO7 using the Magna RIPTM Kit (17-701, Millipore, USA) according to the manufacturer’s instruction. CHIP was performed using the ChIP Assay Kit (P2078, Beyotime, China) following the manufacturer’s instruction.
RNA pull-down
After transfection of biotin-labeled circRILPL1 probes for 24 hours, cells were lysed with the RIP buffer (150 mM KCl, 25 mM Tris-HCl, 0.5 mM DTT, 0.5% NP40) and incubated with 50 ul of Streptavidin Dynabeads (M-280, Invitrogen, USA) overnight at 4°C with rotation. Then the RNA-protein complexes were examined by western blotting.
Immunoprecipitation
Cell lysates were incubated with antibodies (or the control IgG) and 50 ul of protein A/G magnetic beads (Bimake, Houston, Texas, USA) overnight at 4°C with rotation. After washing with GLB+ buffer (10 mM NaCl, 10 mM Tris-HCl, 10 mM EDTA, 0.5% Triton-100) for 3 times, the precipitates were analyzed by western blotting.
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS)
To identify circRILPL1 interacting proteins, cell lysates were incubated with biotin-labeled probes and biotin-affinity magnetic beads overnight at 4°C to pull down circRILPL1-associated proteins. The purified proteins were separated by SDS-PAGE gels and followed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) using Ultimate 3000 RSLC Nano System (Dionex, CA, USA) coupled with LTQ Orbitrap Velos Pro mass spectrometer (Thermo Scientific). Whole proteomic analysis was performed by searching the UniProt KB/Swiss-Prot database using the Proteome Discoverer 1.4 software. An fold change of ≥1.68 or ≤0.62 was used to define differentially expressed proteins.
Animal experiments
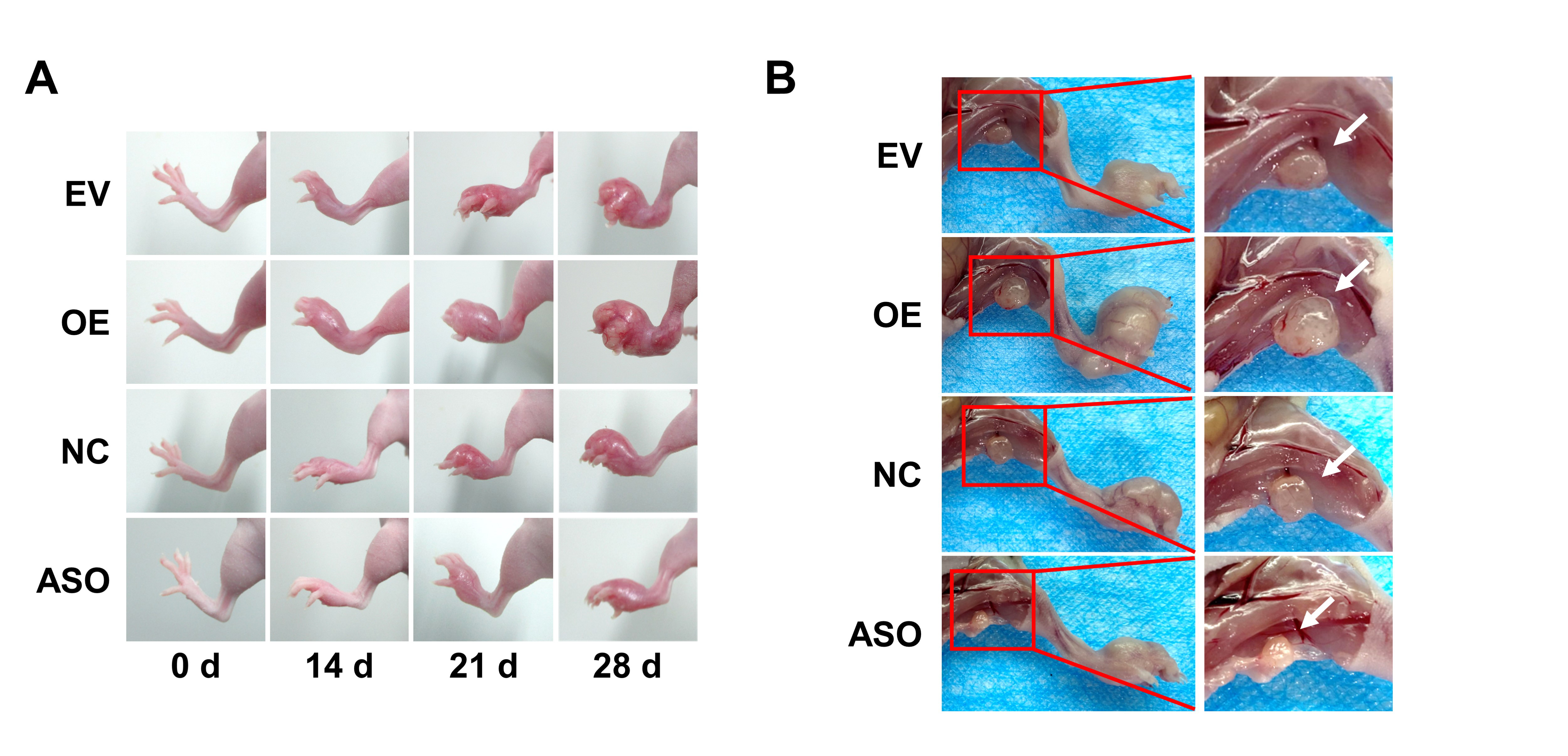
Female BALB/C nude mice were randomly divided into four groups. CNE2 cells (2 × 106) transfected with the vector, the circRILPL1 overexpression plasmid, antisense oligonucleotides against circRILPL1 (ASO-circRILPL1) or the scramble negative control were injected subcutaneously, via tail vein, or footpad, respectively. For the subcutaneous tumor model, mice (6 per group) were sacrificed 30 days after inoculation. The size of the subcutaneous tumor nodule was measured every 5 days and histological examinations were done after 30 days of inoculation. For the tail vein-lung metastasis model, mice were sacrificed 8 weeks after inoculation. The number and area of lung surface metastatic nodules in each mouse were recorded. The lungs were removed, imaged, and embedded in paraffin. Then, the tissues were sectioned for hematoxylin-eosin (H&E) staining, ISH experiments, and metastatic evaluation. For footpad-lymph node metastasis, mice were sacrificed 28 days after inoculation and the ipsilateral inguinal lymph nodes were excised for analysis. All experimental protocols involving animals were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Central South University (Changsha, China).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 8.0. Student's t-test (two-tailed) to compare the difference between two groups of data. All data were represented as mean ± standard deviation (SD). P value < 0.05 was considered statistically significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}