ABCs are induced and maintained following γHV68 infection in a sex-biased manner
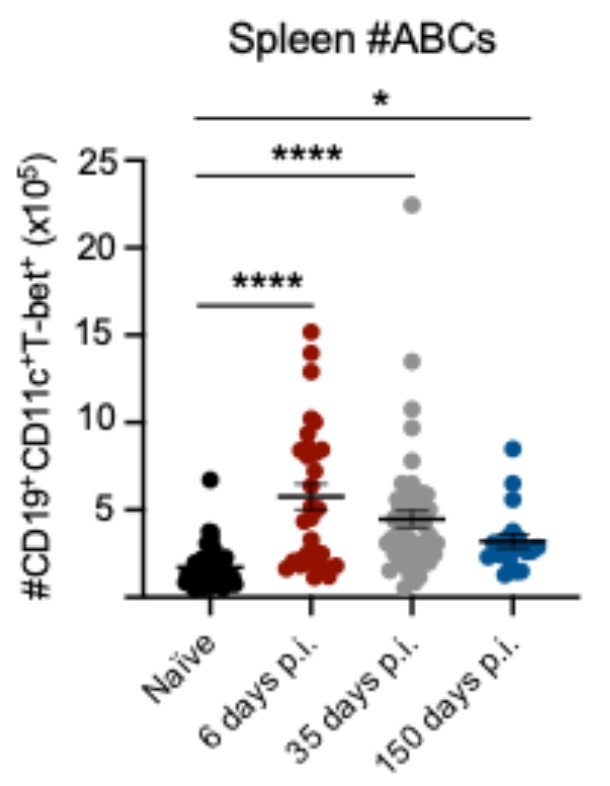
To examine the relative proportion of ABCs during acute and latent γHV68 infection, C57BL/6(J) mice were mock-infected with media (naïve) or infected with γHV68 for 6, 35, and 150 days, blood and spleen were collected and analyzed by flow cytometry to measure ABCs. A gating scheme, described in Fig. 1A, identified CD11c+T-bet+ as a proportion of previously activated B cells (CD19+IgD−). We observed the relative proportion of ABCs increased in circulation during acute γHV68 infection (6 days p.i.) compared to naïve mice (Fig. 1B-C). Once latency was well established (day 35 and 150 p.i.), the proportion of circulating ABCs decreased to pre-infection levels (Fig. 1B-C). This indicates that ABCs circulate in response to the lytic γHV68 infection but do not remain elevated in circulation during latency. In the spleen, the relative proportion and total number of ABCs was increased during acute γHV68 infection (day 6 p.i.) relative to naïve mice and remained elevated during latent γHV68 infection at 35 days p.i. and 150 days p.i. (Fig. 1D-E, Figure S1). These results support previous findings that the spleen is a major site for ABCs following the clearance of acute viral infection9.
Intriguingly, we observed that the proportion of ABCs is increased in females as compared to males in both the blood and spleen throughout infection (Fig. 1F-M). Previously, we demonstrated that ABCs display a sex bias during experimental autoimmune encephalomyelitis and at 35 days post-γHV68 infection in the spleen29. Here, we have extended this finding and shown that ABCs display female sex bias in both the circulation and spleen during lytic and latent γHV68 infection. While ABCs increase in both females and males following γHV68 infection, there is a sex bias leading to greater expansion in females. These results demonstrate that ABCs are increased in circulation during acute γHV68 infection and continue to endure in the spleen long-term during latent infection.
ABCs express anti-viral cytokines in response to latent γHV68 infection
To begin investigating the role of ABCs in the anti-viral response, we measured ABC expression of anti-viral cytokines interferon-γ (IFNγ) and tumor necrosis factor-α (TNF) in the spleen at 6, 35, and 150 days p.i.. We found that during acute γHV68 infection (6 days p.i.),\(\)roughly 40% of ABCs in the spleen expressed IFNγ and TNF, as compared to about 10% of ABCs in naïve mice (Fig. 2A-B). During latent infection (35 days and 150 days p.i.), a significantly increased proportion of ABCs continued to express IFNγ and TNF compared to naïve mice, though the proportion of ABCs expressing IFNγ and TNF was decreased compared to acute infection (Fig. 2A-B). Throughout the acute and latent infection, we observed a downregulation of IL-17A expression on ABCs (Fig. 2C).
To determine if the level of cytokine expression was distinct from other B cells, we compared the proportion of ABCs expressing IFNγ and TNF to non-ABC B cells. Specifically, we examined expression on ABCs compared to CD11c−Tbet+ and CD11c−Tbet− B cells 35 days post-γHV68 infection (Fig. 2D). A significantly increased proportion of ABCs expressed IFNγ and TNF compared to non-ABC B cells, both CD11c−Tbet+ and CD11c−Tbet− B cells, in the spleen (Fig. 2E). The mean fluorescent intensity (MFI) of IFNγ and TNF, of IFNγ or TNF positive cells, was significantly increased on ABCs compared to both non-ABC populations (Figure S2A, B). CD11c−Tbet+ B cells expressed an intermediate level of IFNγ and TNF between ABCs and CD11c−Tbet− B cells (Fig. 2E, Figure S2A, B). Sex differences in ABC cytokine expression were not observed (Fig. 2A-C, Figure S3 A-C). The high level of IFNγ and TNF cytokine expression indicates that ABCs may be functioning in a unique anti-viral capacity during latent infection.
To explore the relationship between latent γHV68 and the ABC population, we infected mice with ACRTA-γHV68, a recombinant strain of γHV68 in which the genes responsible for latency are deleted and a lytic gene, RTA, is constitutively expressed30. As a result, γHV68 infection is cleared following acute infection without ever establishing latency. We found that, at 35 days p.i., a time point at which ACRTA-γHV68 is cleared, ABCs are increased to the same level in the spleens of ACRTA-γHV68 infected mice as compared to spleens in mice infected with WT γHV68 (Fig. 2F). Thus, ABCs persist in the absence of latent virus, in a manner similar to memory cells that remain following acute infection.
While the proportion of ABCs remained elevated in the absence of latent virus, cytokine expression was altered. The proportion of ABCs in ACRTA-γHV68-infected mice expressing IFNγ and TNF at 35 days p.i. was significantly reduced compared to those infected with WT γHV68 while remaining significantly elevated compared to naïve mice (Fig. 2G-H). Additionally, the IFNγ+ ABCs in mice infected with ACRTA-γHV68 displayed a lower MFI than those from γHV68-infected mice, though the TNF MFI was not significantly different between the two groups (Figure S2C, D). Without the presence of the latent virus, we observe a decrease specifically in the proportion of ABCs that doubly express both IFNγ and TNF as well as an increase in ABCs that are negative for the expression of either IFNγ and TNF as compared to ABCs from WT γHV68-infected mice (Fig. 2I). This finding indicates that ABCs respond to the latent virus by producing both IFNγ and TNF.
Together, these results show that ABCs are a predominant B cell subset expressing anti-viral cytokines IFNγ and TNF during latent γHV68. Further, our data establish that high levels of expression of these cytokines are dependent on the presence of the latent virus. These findings suggest that ABCs recognize the presence of the quiescent latent virus and respond by expressing anti-viral cytokines.
ABCs are susceptible to γHV68 infection but are not a major viral reservoir
γHV68 is known to infect B cells, in particular germinal centre and memory B cells31 − 34. To determine if ABCs are directly infected by γHV68, we used a previously developed fluorescent strain of γHV68, γHV68.H2bYFP35 where fluorescence is easily detectable during acute infection, but over time falls off during latent infection. Mice were infected with γHV68.H2bYFP and 8 days p.i. spleens were collected, and flow cytometry was performed. We found that, during acute infection, a small proportion of ABCs were infected with γHV68. The same proportion of ABCs were positive for the fluorescent virus (1.2 ± 0.3%) as observed in previously activated B cells (1.1 ± 0.2%, Fig. 3A-B), demonstrating that ABCs are not preferably targeted for infection. To our knowledge this is the first evidence of a virus directly infecting ABCs. We next asked what proportion of the γHV68 reservoir is made up of ABCs. The primary reservoir for γHV68 is previously activated B cells which aligns with our results that 73% of γHV68-infected cells are IgD− B cells (Fig. 3C). We found that ABCs make up only 6% of γHV68-infected cells (Fig. 3C). Other infected cell populations included innate cells such as macrophages and DCs, in alignment with previous findings31, as well as a small proportion of NK and T cells (Fig. 3C). We also examined the relationship between infection of ABCs and expression of IFNγ. We observed that during acute infection the proportion of ABCs expressing IFNγ was significantly lower in ABCs infected with γV68 (4.3\(\pm\)1.3 %) as compared to uninfected ABCs (32.2\(\pm\)1.0%, Fig. 3D). These results indicate that direct infection of ABCs was not driving IFNγ production and that at least during acute infection, ABCs are susceptible to virus-driven downregulation of IFNγ. These findings demonstrate that although a portion of ABCs are infected with γHV68, ABCs are not the major target for γHV68.
ABCs are dispensable for the control of acute infection and establishment of latency
To further examine the role(s) of ABCs in γHV68 infection, mice with a floxed B cell specific T-bet deletion were followed post-infection. Specifically, Tbx21fl/flCd19cre/+ (KO) and littermate Tbx21fl/flCd19+/+ (Ctrl) mice were infected with γHV68 for 6 or 35 days and the spleen was collected to examine the quantity of γHV68 by qPCR and the immune cell composition (Fig. 4A). Mice were genotyped by PCR and loss of ABCs in KO mice was confirmed by flow cytometry (Fig. 4B). No differences in clinical symptoms or weight changes were observed during γHV68 infection between Ctrl or KO mice (Figure S4A).
To determine if knocking out ABCs alters immune cell composition, Ctrl and KO mice were infected or mock-infected with γHV68 and spleens analyzed at 6 or 35 days by flow cytometry to examine various T cell, B cell, and innate immune cell populations. No difference in the total number of splenocytes was observed (Figure S4B). The composition of the splenic immune profile was similar between Ctrl and KO mice with no significant differences observed in the relative proportions of B cells, CD8 T cells, DCs, neutrophils, NK cells, or macrophages between mock-infected mice or those infected with γHV68 for 6 or 35 days, though KO mice had greater proportions of CD4 T cells than Ctrl mice 35 days post-infection (Figure S5).
We then measured the quantity of γHV68 in the spleens of Ctrl and KO mice infected for 6 and 35 days by qPCR. We found no difference in that the quantity of γHV68 at 6 and 35 days p.i. between Ctrl and KO mice (Fig. 4C-D) and the quantity of γHV68 did not differ between male and female mice (Figure S4C-D). To confirm that KO mice effectively control the lytic infection and develop latency, we infected Ctrl and KO mice with latency-deficient ACRTA-γHV68 for 35 days. We found that KO mice, like Ctrl mice, displayed similar viral loads that were near or below the limit of detection when infected with ACRTA-γHV68 (Fig. 4E). As no virus was detected at day 35, mice lacking ABCs were effectively controlling the lytic infection. Furthermore, no differences were observed in the relative expression of viral genes associated with lytic infection (Orf50, Orf68) or latent infection (Orf73) between Ctrl and KO mice (Fig. 4F). Orf50 encodes the replication and transcription activator protein, which initiates viral lytic gene expression36. Orf68 encodes a packaging protein that assists in moving the newly replicated viral genomes to the packaging motor, where they are loaded into capsids37. Orf73 encodes for the latency-associated nuclear antigen that is required for the establishment and maintenance of latent infection38,39. As such, equal expression of lytic and latency-associated genes Orf50, Orf68, and Orf73 between Ctrl and KO mice suggests that Ctrl and KO mice harbor similar levels of lytic and latent γHV68. Together, these results demonstrate that ABCs are not required for the clearance of acute infection and establishment of latency in steady-state conditions.
Lack of ABCs results in a dysregulated γHV68 antibody response but does not alter the viral reservoir or anti-viral T cell response
ABCs are known to secrete anti-viral IgG2a/c6, and transfer of serum into mice without ABCs during LCMV infection was partially able to restore control of the infection15. To examine the antibody response in Ctrl versus KO mice, mice were infected with γHV68 for 35 days and sera was collected. The levels of anti-γHV68 IgG were the identical between Ctrl and KO mice, though Th1-associated IgG2c was significantly decreased in KO mice, while Th2-associated IgG1 was significantly elevated, when compared to Ctrl mice (Fig. 5A-C). These results suggest that ABCs are the primary secreters of anti-γHV68 IgG2, though in their absence there is compensation by other B cell subsets resulting in increased anti-γHV68 IgG1 antibodies.
We next asked if the γHV68 reservoir was altered in mice without ABCs, as a portion of γHV68-infected cells were ABCs. In particular, we posited that an altered viral reservoir could impact the ability of KO mice to control γHV68 latency; previous findings indicate that various cell types infected by γHV68 may display differential susceptibility to reactivation23,40. To compare the viral reservoir in Ctrl and KO mice, we infected mice with a fluorescent strain, γHV68.H2bYFP, and, at 8 days p.i., examined immune cell populations that comprise the γHV68-infected population. We observed no difference in the proportion of infected cell populations, including T cells, DCs, macrophages, NK cells, and naïve and previously activated B cells (Fig. 5D). This suggests that the cell populations infected with γHV68 are not substantially altered in KO mice compared to Ctrl mice, making it unlikely that changes to the reservoir impacted susceptibility to viral reactivation.
To ask whether ABCs influence anti-viral immune cells in the spleen, we next examined three cell populations previously shown to be important for control of latent γHV68: IFNγ-producing cells, γHV68-specific CD8+ T cells, and Vβ4+CD8+ T cells. IFNγ is present at low levels during latency41 and is critical for controlling γHV68 reactivation from latency23,24,42. In particular, IFNγ-producing T cells are known to block γHV68 reactivation23,43−45. CD8+ T cells that harbor the Vβ4 TCR were also examined. CD8+Vβ4+ T cells expand following γHV68 infection and reach their highest levels during latency, wherein they persist throughout infection without taking on an exhausted phenotype46 − 48. γHV68-specific CD8+ T cells were also measured with tetramers to p79, an immunodominant γHV68 epitope for which CD8-specific memory T cells remain throughout latent infection49. We observed that the proportion of CD4+ and CD8+ T cells that express IFNγ were unchanged between mice with and without ABCs (Fig. 5E-F). Further, no differences were observed in the proportion of either Vβ4+CD8+ T cells or γHV68 p79-specific CD8+ T cells between mice with and without ABC circulating populations (Fig. 5G-H). Additionally, we found that the proportion of NK cells and B cells that express IFNγ, and the MFI of IFNγ on these populations, was not changed between Ctrl and KO mice whether mock-infected or infected for 6 or 35 days (Figure S6). That a similar proportion of B cells expressed IFNγ indicates that another B cell subset likely compensates for the loss of IFNγ-expressing ABCs. Collectively, these findings indicate that ABCs are not acting to stimulate anti-viral immune cell populations in the spleen.
Together, these data indicate that knocking out ABCs leads to dysregulation of the γHV68 antibody response, without altering the γHV68 reservoir or T cell populations responding to the latent virus.
In the absence of ABCs, reactivation of γHV68 occurs more readily following infections
We posited that the persistence of ABCs long-term during γHV68 and their secretion of IFNγ, TNF, and anti-viral antibodies suggest that ABCs have an important role in the suppression of latent virus reactivation. To examine this role, ex vivo reactivation assays were performed on splenocytes from Ctrl and KO mice at 35 days p.i.. Splenocytes from γHV68-infected KO mice demonstrated more frequent virus reactivation in culture compared to splenocytes from γHV68-infected Ctrl mice (Fig. 6A). This finding indicates that there is an increased propensity for γHV68 reactivation in the absence of ABCs, although the assay does not disentangle whether infected cells have an altered cell-intrinsic susceptibility to reactivation or if there is a cell-extrinsic influence on reactivation.
To further interrogate the role of ABCs in suppressing γHV68 reactivation, we next asked if mice without ABCs are more susceptible to γHV68 reactivation following infectious challenge. Importantly, we chose to examine the response to viruses that do not typically reactivate γHV68,50 such as LCMV and coxsackievirus B4 (CVB4). These heterologous viral infections were performed as physiological mimics to ask if they were sufficient to cause reactivation of latent γHV68 in the absence of ABCs. Specifically, KO and Ctrl mice were infected with γHV68 for 35 days and then challenged with LCMV or CVB4 (Fig. 6B). No clinical symptoms were observed during either of the challenges in Ctrl or KO mice and there was no difference in the relative quantity of LCMV and CVB4 in the spleens of Ctrl versus KO mice (Fig. 6C-D). Following challenge with LCMV or CVB4, mice lacking ABCs had elevated quantities of γHV68 in the spleen compared to mice with ABCs (Fig. 6E-F). We reasoned that the increased γHV68 load following challenge may be due to increased reactivation and, in support, observed increased relative expression of two lytic-associated γHV68 genes, Orf50 and Orf68, in KO mice compared to Ctrl mice following challenge with the heterologous viruses (Fig. 6G-H). Alternatively, we did not observe a difference in expression of the latency-associated gene Orf73, between Ctrl and KO mice (Fig. 6I). This also indicates that it is not simply an expansion in the number of latently infected B cells in the spleen and is a change in the active lytic expression of more virus. Further, we did not observe significant cell number increases in B cells between the KO and Ctrl mice post challenge (Figure S7). These findings make clear that ABCs act to impede latent γHV68 reactivation in the face of mild stresses such as heterologous infection.
{kind=link}