3.1. General techniques
Melting points were determined on a Stuart SMP30 melting point apparatus. IR spectra (KBr) were recorded on a JASCO 6100 spectrophotometer. NMR spectra were recorded on a JEOL AS 500 (DMSO-d6, 1H: 500 MHz, 13C: 125 MHz) spectrometer, JEOL USA, Inc. Mass spectra were recorded on a Shimadzu GCMS-QP 1000 EX (EI, 70 eV) spectrometer, Shimadzu corporation, Kyoto, Japan. Elemental micro analyses were carried out utilizing a Vario Elemental analyzer, Elementar Analysensysteme GmbH, Langenselbold, Germany. Figs. S1–S4 of Supplementary material show the spectral data. The starting compound 2-(chlorocarbonyl)phenyl acetate (1) was prepared according to the previously reported procedures [28, 29].
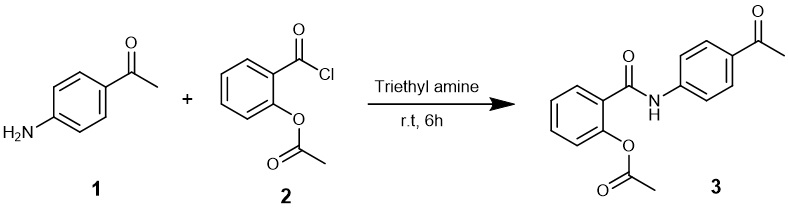
3.2. Synthesis of 2-(4-Acetylphenylcarbamoyl)phenyl acetate (3)
To a stirred solution of 4`-aminoacetophenone (1) (1.35 gm, 10 mmol) and N,N-diethylethanamine (1.48 ml, 11 mmol) in dichloromethane 25 ml, was added 2-(chlorocarbonyl)phenyl acetate (2) (1.98 gm, 10 mmol) portion-wise over 30 min. and the mixture was allowed to be stirred at room temperature for 6 hrs. The mixture was filtered, the solvent evaporated under reduced pressure, and then the solid obtained was washed with water, dried and recrystallized from benzene/pet. ether 60–80.
Buff crystals; yield (2.65 gm) 89%; mp 151–153°C; IR (νmax/cm− 1): 3297 (NH), 1659,1679, 1760 (C = O); 1H NMR (DMSOd6) δ (ppm): 2.18 (s, 3H, CH3), 2.52 (s, 3H, COCH3), 7.24, 7.26 (dd, 1H, J = 1.20, 1.10 Hz, CH), 7.39 (t, 1H, J = 8.13 Hz, CH), 7.57 (t, 1H, J = 8.65 Hz, CH), 7.70, 7.71 (dd, 1H, J = 1.65, 1.65 Hz, CH), 7.85 (d, 2H, J = 8.8 Hz, CH), 7.94 (d, 2H, J = 8.8 Hz, CH), 10.71 (s, 1H, NH); 13C NMR (DMSO-d6) δ (ppm) 20.84, 26.60, 119.28, 123.48, 126.08, 129.37, 129.45, 129.53, 129.57, 132.03, 132.32, 143.64, 148.30, 164.80, 169.04, 196.74; MS: m/z (%) 297.88 (M+, 9.97); Anal. calcd. for C17H15NO4 (297.31): C, 68.68; H, 5.09; N, 4.71. Found: C, 68.66; H, 5.10; N, 4.70.
{kind=link}