Prion diseased mice show signs of neuroinflammation prior to neuronal dysfunction.
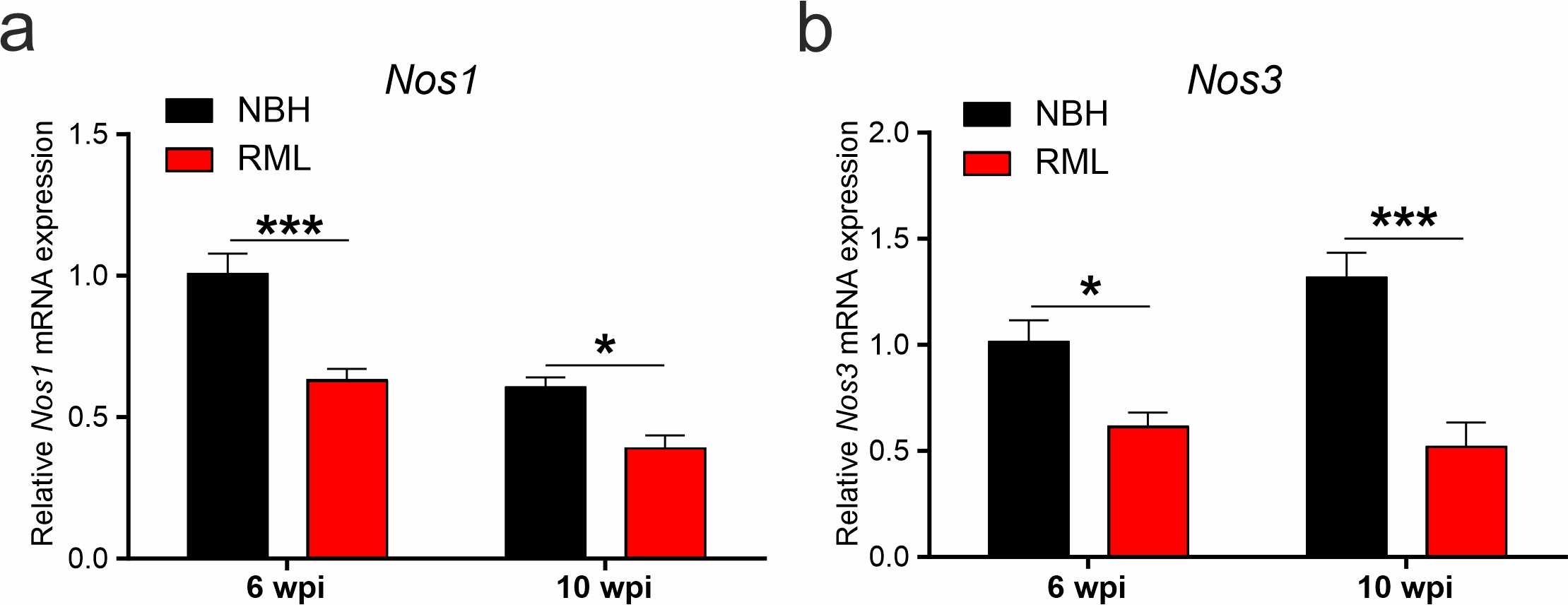
Previously we have found that in the hippocampus and cortex of prion infected mice the neuronal metabolism, not only related to the NO signalling cascade, is aberrantly regulated at 10 w.p.i. [8]. At this advanced time point we found evidence for numerous pathological markers such as high levels of misfolded prion protein (PrPSc) in the hippocampus and cortex, enhanced glial fibrillary acidic protein (GFAP) expression associated with nitrergic and oxidative stress, and hippocampal neuronal loss as well as learning and memory deficits [37, 41]. However, as the disease is highly progressed by 10 w.p.i., here we investigated earlier disease-relevant pathways which may enhance a potential rescue of neurodegeneration. We examined the contributions of neuroinflammatory and oxidative stress-related signalling in prion infected mice (RML) at a time point prior to onset of reported disease symptoms (at 6 w.p.i.), as well as advanced stage disease (at 9-10 w.p.i.). We first tested gene expression for the inflammatory suppressor of NF-kB signalling, Ikbα, and found it to be downregulated at 6 w.p.i. (P=0.038, two-way ANOVA, n=4 mice each, Fig. 1a). RNA expression for the peroxisome proliferator-activated receptor-gamma coactivator, PGC-1α, which controls the expression of genes related to the generation of ROS and prevents oxidative stress by reducing the production of ROS [42], is also downregulated at this early stage of disease (P=0.0015, two-way ANOVA, n=4 mice each, Fig. 1c), as is the prion-interacting protein stress-inducible phosphoprotein 1, STIP1 [43] (P=0.0013, two-way ANOVA, n=4 mice each, Fig. 1d). At 10 w.p.i. we found a strong increase in mRNA levels for Ikba (P=0.012) as well as NCF1 (P<0.0001), one of the components of NADPH oxidase (p47-phox, Fig. 1a, b, n=4 mice each). Furthermore, PGC-1a (P<0.0001, two-way ANOVA) and STIP1 (P=0.0047, two-way ANOVA) mRNA levels remain reduced at this later disease stage (Fig. 1c, d). Importantly, Nos2 mRNA (iNOS) is increased at 10 w.p.i. (P=0.0266, two-way ANOVA, Fig. 1e) with Nos1 (nNOS) and Nos3 (eNOS) mRNA levels being strongly reduced at 6 and 10 w.p.i. (Additional file 1: Figure S1). The enhanced iNOS activity is likely a result of activated astroglia signaling which we further illustrated using the NADPH diaphorase assay at 6-12 w.p.i. (Fig. 1f). We have previously shown that NBH mice lack any significant NADPH diaphorase signals at 10 w.p.i. [8]. This data illustrates a strong increase of NOS (NADPH activity) from 8 w.p.i. onwards in RML mice with signals reminiscent of activated astroglia in the CA1 region spreading to the whole hippocampus at 10 and 12 w.p.i.. Further characteristics for upregulated astroglia were detected by immunostaining for GFAP which showed a significantly stronger expression at 10 w.p.i. in RML hippocampi (P=0.045, Student’s t-test, Fig. 1g). Altogether our data indicate that an early onset neuroinflammatory contribution to prion disease is detectible from 8 weeks onwards. Thus our approach to start targeting NO signaling will focus on time points following 6 w.p.i. onwards.
Rescue of functional neuronal decline in neurodegeneration by reducing nitrergic stress.
We asked whether interference with neuroinflammatory NO signalling prior to detectable prion misfolding might alleviate some of the neuronal symptoms reported in the disease. In order to address this question, subsequent studies only focussed on RML mice and the effects of treatment since we did not find any indications for aberrant nitrergic signalling in NBH controls (Fig. 1). We quantified the function of CA1 pyramidal neurons and their Schaffer collateral-mediated synaptic inputs as a read-out of neuronal health in RML prion-infected mice treated daily from 6 w.p.i. with the NOS inhibitor, L-NAME in comparison to age-matched vehicle RML controls. Both cohorts were assessed by electrophysiology at 7 and 9 w.p.i.. These time points were chosen to study the effects of NOS manipulation based on data presented in Figure 1 at which we expect pathological neuroinflammation to become dominant. Fig. 2a shows representative Schaffer collateral-evoked excitatory postsynaptic currents (eEPSC) in pyramidal CA1 neurons in RML mice at 7 and 9 w.p.i. following stimulation by a 30 Hz train. The recordings illustrate a vast deterioration in neuronal function by 9 w.p.i.. Mean train eEPSC amplitudes from vehicle (RML) or L-NAME treated mice are summarised in Fig. 2b, c (RML, black; +L-NAME, red, n=9 mice each).
Following 3 weeks of daily treatment with L-NAME, mean initial current amplitudes were increased at 9 w.p.i. compared to RML vehicle control mice (P=0.029, two-way ANOVA, Fig. 2d). Cumulative postsynaptic current analysis (Fig. 2e, f), a measure to estimate available vesicle pool sizes based on the cumulative eEPSC amplitude plots, revealed a strong recovery of the vesicular pool size at 9 w.p.i. following L-NAME treatment (P=0.043, two-way ANOVA, Fig. 2g) suggesting an improvement of synapse function. An important regulatory mechanism for synaptic release is the control of release probabilities. As a measure to determine the initial synaptic release probabilities we assessed paired-pulse ratios (PPR, eEPSC2/eEPSC1 amplitudes) at 33 ms inter-spike intervals of Schaffer collateral-stimulated eEPSCs in diseased and treated mice. PPR values of synaptic responses in hippocampi from diseased mice revealed a significant increase at 9 w.p.i. indicating a reduction in release probability over time (P=0.0145, two-way ANOVA, Fig. 2h). L-NAME treatment, however, suppressed this increase in PPRs, which suggests that NOS inhibition prevents different aspects of synapse dysfunction (Fig. 2h). A further assessment criterion of neuronal health is the ability of synapses to spontaneously release vesicles. These spontaneous or miniature EPSCs (mEPSC) provide important signals for physiological neuronal homeostatic signaling [44]. Consistent with a general neuronal dysfunction in prion disease, we found that miniature EPSC (mEPSC) amplitudes and spontaneous firing frequencies were reduced at 9 w.p.i. (Fig. 3a-c). Analyzing the relative cumulative distributions of mEPSC amplitudes and inter-spike-intervals (ISI) revealed a strong leftward shift in amplitudes and rightward shift in ISI in prion disease at 9 w.p.i. compared to 6 w.p.i. (Fig. 3b, c, amplitudes: P<0.0001, D=0.571; ISI: P<0.0001, D=0.738, Kolmogorov-Smirnov test: 9 vs 6 w.p.i.) confirming again compromised neurotransmission. A protective effect of L-NAME treatment was observed at 9 w.p.i. (Fig. 3b, c, amplitudes: P<0.0001, D=0.571; ISI: P<0.0001, D=0.683, Kolmogorov-Smirnov test: 9 w.p.i. vs 9 w.p.i. + L-NAME treatment). The effects of prion disease are further illustrated for mean mEPSC amplitudes and frequencies (Fig. 3d, e, black, P<0.05, one-way ANOVA) and importantly, L-NAME treatment leads to recovery of both parameters over the time course of the disease (Fig. 3d, e, red, P<0.05, one-way ANOVA).
We also assessed neuronal function by measuring the ability of CA1 pyramidal neurons to generate current-evoked action potentials (AP), and characterized the underlying whole-cell potassium and sodium currents. Both, the ion channel functions and AP propagation play crucial roles in regulating neuronal performance and allow the neurons to sustain information transmission. AP parameters such as half width and amplitude are main characteristics, both predominately determined by potassium and sodium channel activities, respectively. L-NAME treatment led to a reduction of the increased AP half-width at 9 w.p.i. indicating an augmentation of potassium currents (P=0.002, one-way ANOVA). AP amplitudes were reduced at 7 w.p.i. (P=0.271, one-way ANOVA) and increased at 9 w.p.i. (P=0.018, one-way ANOVA) following L-NAME treatment (Fig. 4a, b). As both measures are determined by the activities of voltage-gated potassium and sodium channels, we directly assessed both, potassium and sodium currents by voltage-clamp. Current-Voltage (IV) relationships show a strong decline of outward potassium currents (Fig. 4c, d) and inward sodium currents (Fig. 4e) at 9 w.p.i. confirming the observed changes in AP parameters, with L-NAME treatment recovering both currents substantially at 9 w.p.i., substantiating the AP data at 9 w.p.i. and confirming a neuroprotective role of L-NAME treatment. The data on neuronal physiology were supported by measurements of levels of neuronal protein involved in synaptic function and neuronal fidelity. Protein levels of several synaptic markers, including MUNC 18, SNAP-25 and complexin 1/2 were reduced with others showing strong tendencies of lower expression (synaptophysin, synaptobrevin, synapsin) at 9 w.p.i. (Additional file 2: Figure S2). Protein expression of the voltage-gated potassium channel Kv3.1, the main conductance contributing to the IV relationships in Fig. 4c, d and determinant of AP half width, shows a decline at 9 w.p.i.. Altogether, the data illustrate the broad neuronal dysfunction at later disease stages and the potent effects of L-NAME treatment to recover several aspects of neuronal dysfunction by suppressing excessive NO signaling in vivo.
Protein 3-nitrotyrosination is driven by oxidative stress and enhances protein glycation in prion disease.
Protein 3-nitrotyrosination is characteristic of protein-misfolding neurodegeneration [22, 45]. 3-NT occurs under conditions of oxidative stress and aberrant nitrergic activity. We confirmed a decreased ratio of reduced to oxidized glutathione GSH/GSSG in prion diseased mice at 9 w.p.i. (P=0.0039, control NBH vs RML mice, Student’s t-test, Fig. 5a) providing evidence for enhanced oxidative stress as one hallmark of the disease and confirming previously reported increases in oxidative stress levels in prion-diseased mice [8]. Together with the enhanced levels of nitrergic stress associated with augmented expression levels of iNOS mRNA (Fig. 1), we found total amounts of 3-nitrotyrosinated proteins to be >5-fold elevated in prion infected mice compared to NBH controls at 9 w.p.i. (P=0.0005, control NBH vs RML, one-way ANOVA, Fig. 5b). Importantly, following treatment with the NOS inhibitor L-NAME, these elevated levels of 3-NT could be reversed back to control values (P=0.0005, RML+L-NAME vs RML, one-way ANOVA, Fig. 5b).
Both AD and CJD are associated with neurotoxic glycation signalling as well as a strong presence of AGEs [22, 23, 36]. AGEs are produced by the non-enzymatic glycation of amino compounds with reducing sugars through a series of sequential and irreversible reactions. The predominant neuronal precursor to induce protein glycation is DHAP which spontaneously forms methylglyoxal, a strong glycating agent [46]. We have measured DHAP by mass spectrometry and found levels to be ~2-fold increased (P=0.0027, Student’s t-test, Fig. 5c) in the hippocampus of prion-infected mice. As TPI is the upstream enzyme in the cascade to produce DHAP, a reduction of its enzymatic activity would be responsible for the observed changes in the levels of its metabolites. It has been previously reported that mutations of TPI carrying the tyrosine substitutions Y165F and Y209F, both of which are located at the catalytic center, render this enzyme inefficient when expressed in human neuroblastoma cells thereby shifting the balance between GAP and DHAP towards the latter and reducing cell viability [23]. Based on these findings, we tested whether we could detect a nitrergic post-translational modification of TPI in prion-diseased mice that would ultimately be responsible for an accumulation of DHAP. Indeed, when performing immunoblotting for 3-nitrotyrosination after having immuno-precipitated TPI, we detected an increase of 3-NT TPI protein levels in disease (P=0.033, one-way ANOVA, RML vs NBH, Fig. 5d). Importantly, treatment with L-NAME prevented the increase in 3-NT TPI levels and reversed it back to control levels (P=0.484, one-way ANOVA, NBH vs RML+L-NAME, Fig. 5d). Total amounts of TPI protein were unchanged (P=0.163, one-way ANOVA, Fig. 5f). The production of AGE following TPI nitrotyrosination is directly linked to the activation of the receptor for AGE (RAGE) and the expression of RAGE reflects a positive confirmation for AGE signaling [47]. To test whether we could confirm AGE signaling in prion mice we examined RAGE expression and found that prion diseased mice exhibited markedly enhanced levels of RAGE (P=0.0074, one-way ANOVA, Fig. 5e). We tested the effects of NOS inhibition on RAGE expression and found that treatment with L-NAME ameliorated these increases (P=0.0243, one-way ANOVA, Fig. 5e, note the lower band represents actin) providing strong evidence for the NO-mediated 3-NT/TPI/DHAP/RAGE signaling cascade in prion disease which signifies a disease-associated neurotoxic pathway.
Nitric oxide and glycation signalling impact on prion misfolding.
The pathological scrapie form of the prion protein is heavily glycated resulting in mainly Nε-(carboxymethyl)lysines at up to eight lysine residues, whereas PrPC does not contain glycated amino acids [48, 49]. It has been shown that mono- or di-glycation affects the molecular weight of the prion protein resulting in multiple bands following immunoblotting. We thus tested for expression of prion protein in hippocampal tissues. Importantly, we noticed the occurrence of bands above the expected PrPc molecular weight (~36 kDa) in hippocampal samples of RML mice at 9 w.p.i. detected using the ICSM35 antibody raised to specifically recognise β-sheet-rich structures within prion proteins [50, 51] (Fig. 6a). These higher weight bands might therefore reflect glycated and β-sheet-enriched prion protein.
We next assessed hippocampal tissue by immunocytochemistry and stained for prion protein to investigate the localisation of the misfolded protein in NBH and RML mice with and without L-NAME treatment. In order to specifically visualise β-sheet-rich PrPSc that is prerequisite for prion aggregation we immunostained samples using the ICSM35 antibody [50, 51]. We found that prion signals were almost exclusively detected in the cytosol of cubiculum neurons in RML mice reminiscent of possible protein aggregates formed within the cytosol [49, 52] (Fig 6b, c). Cubiculum areas from NBH mice did not show any labelling in neurons (Fig. 6b) confirming PrPSc specific detection in RML mice (note also the strong loss of DAPI signals in RML samples indicative of substantial neuronal loss which was reversed after L-NAME treatment, Fig. 6b). Following counting of ICSM35-positive (PrPSc) cells in the hippocampus, we found larger proportions of cells in RML mice compared to RML mice receiving L-NAME treatment suggesting a reduction in the occurrence of prion protein misfolding (RML: 38.8 ± 4.1%, RML+L-NAME: 23.4 ± 2.7%, total cell number: 679, P=0.0346, Student’s t-test, [n=5 RML mice, n=3 RML+L-NAME treated mice], Fig. 6b, c). These data therefore indicate that reducing nitrergic stress could slow down the progression of protein misfolding and/or aggregation. We further noticed that the absolute numbers of pyramidal neurons in the hippocampus of L-NAME treated mice was similar from control NBH hippocampi whereas vehicle treated RML mice showed a significant reduction in cell numbers (NeuN positive cells per mm2: NBH: 0.0021 ± 0.0001 (n=3 mice); RML: 0.0007 ± 0.0001 (n=5 mice), P=0.0324 vs NBH; RML+L-NAME: 0.0013 ± 0.0003 (n=3 mice), one-way ANOVA; Fig. 6b, d (representative images of NeuN and DAPI labelled tissues). This data suggest that L-NAME treatment results in a reduction of neuronal loss consistent with the L-NAME mediated rescue of neuronal function at this time point of disease (Fig. 2-4).
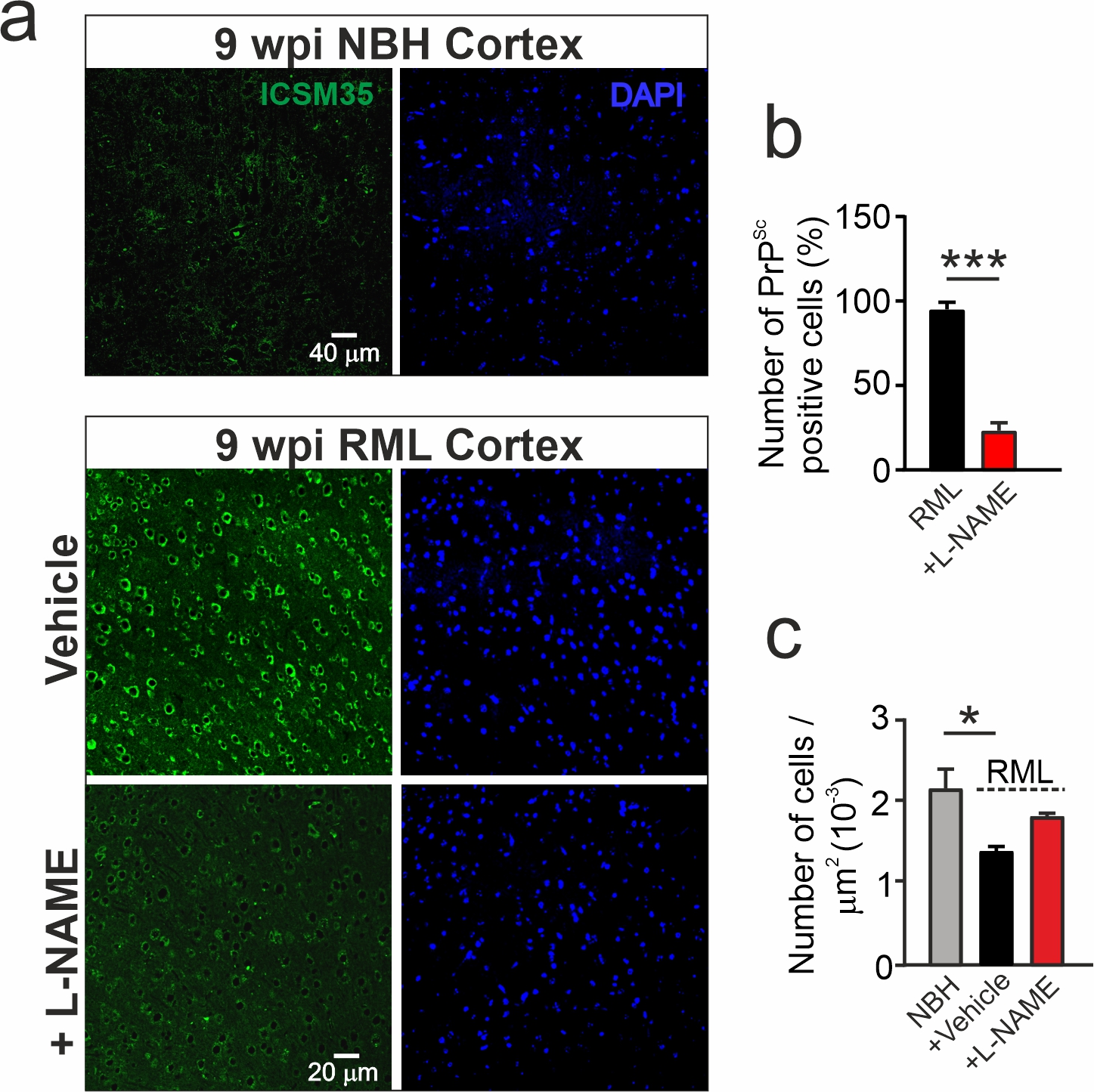
Previously we reported that the metabolome of the hippocampus as well as the cortex is altered in prion disease, with both regions showing significant increases in nitrergic and oxidative stress signalling at 10 w.p.i. [8]. Furthermore, diseased mice exhibit behavioural deficiencies in burrowing activity at 9 w.p.i. [37], a behaviour associated with hippocampal but also cortical dysfunction [53]. In parallel to the hippocampal increases in DHAP levels, we also confirmed enhanced levels of DHAP in the cortex of RML mice (scaled intensity: NBH: 0.81 ± 0.07, n=8; RML: 1.07 ± 0.07, n=8; P=0.023, Student’s t-test, data not shown) suggesting a similar upregulated glycation signaling which could underlie the observed prion misfolding. To test if we can detect potential signs of prion misfolding in the cortex as well and beneficial effects of L-NAME treatment in cortical regions at 9 w.p.i. we assessed cortical tissues for misfolded prion protein by immunocytochemistry. We found that in the cortex from RML mice prion protein positive signals exhibit clusters within the cytosol showing typical punctate form of localisation pattern as seen in the immunocytochemistry data in the hippocampus reflecting misfolding of the protein which was absent in tissue from NBH mice (Additional file 3: Figure S3a). Following counting of cells exhibiting such PrPSc clusters, we found that RML samples exhibited high numbers which were greatly reduced following L-NAME treatment suggesting a reduction of neurons possessing b-sheet-rich prion protein (RML: 95.0 ± 2.2%, RML+L-NAME: 24.2 ± 5.1%, total cell number: 1892, P<0.0001, Student’s t-test, [n=5 RML mice, n=3 RML+L-NAME treated mice], Additional file 3: Figure S3b). Importantly, we also assessed the total number of cortical neurons and found that L-NAME treatment reversed the reduction in cell loss caused by disease (cells per mm2: NBH: 0.0022 ± 0.0002 (n=3 mice); RML: 0.0014 ± 0.00006 (n=5 mice), P=0.0119 vs NBH; RML+L-NAME: 0.0018 ± 0.00004 (n=3 mice), one-way ANOVA; Additional file 3: Figure S3a, c).
Together, our data provide evidence that in vivo NOS inhibition for 3 weeks can reduce the amount of prion protein misfolding which positively impacts on neuronal health. We propose that nitrergic stress exerts its effects via post-translational modification, and associated inhibition, of TPI leading to an accumulation of glycated proteins. The resulting generation of advanced glycation end products may impact on prion protein misfolding as AGE-assisted conversion of PrPC into PrPSc itself as it is reported for α-synuclein [26, 27] or Ab [31].
{kind=link}
{kind=link}