Materials
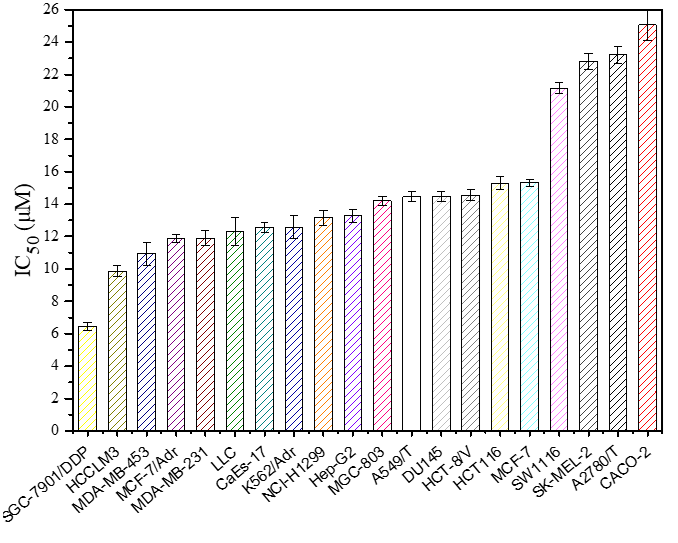
Morusin (Chengdu Ruifensi BioTech Co., Ltd., China; Purity≥98%) MCF-7, A549/T, CaEs-17, HePG2, HCCLM3, A2780/T, LLC, SGC-7901/DDP, MDA-MB-231, MCF-7/Adr, MGC-803, HCT-116, NCI-H1299, DU145, SK-MEL-2, SW1116, K562/Adr, CACO-2, HCT-8/V, MDA-MB-453 (Nanjing KeyGen Biotech Co., Ltd., China)
Cell culture
Three breast cancer cells of MCF-7, MDA-MB-453 and MDA-MB-231, human oesophageal cancer cell of CaEs-17, gastric cancer cell of MGC-803, human lung cancer cell of NCI-H1299 and colorectal cancer cell of SW1116 were subcultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with heat inactivated FBS (foetal bovine serum, 10%), penicillin (1%) and streptomycin. These cells were maintained at 37°C in a 5% CO2 incubator. The cells of MDA-MB-453 were suspended, and the others were adherent cells. Culture solution of adherent cells in the process of passage, with PBS flushing one time, and circular shrinkage was observed in microscope and joined medium to stop trypsin digestion. Then, the solution was centrifuged for 5 min (1000 rpm/min) and made cells sedimentation, joined new culture solution, blowed into the cell suspension gently and inoculated cells respectively.
Human hepatoma cell HCCLM3, mouse lung cancer cell LLC and human colon cancer cells HCT-116 and CACO-2 were subcultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing heat inactivated 10% FBS, 1% penicillin and streptomycin. These cells were cultured at 37°C in a 5% CO2 incubator. LLC was the hemiparietal cell, and the other cells were adherent. The culture process was the same as that described above.
Human hepatoma cell (Hep-G2), human prostatic cancer cell (DU145) and human malignant melanoma cell (SK-MEL-2) were subcultured in extreme magnetoelectric (EME) medium supplemented with heat inactivated FBS (10%), 1% penicillin and streptomycin. Cells were maintained at 37°C in a 5% CO2 incubator. All cells were adherent growth cells, and the culture process was the same as that described above.
Human lung cancer A549/Taxol cells, human ovarian cancer A2780/Taxol cells, human gastric cancer SGC-7901/DDP cells, human breast cancer MCF-7/Adr cells, human leukaemia K562/Adr cells and human colorectal HCT-8/V cells were subcultured in RPMI-1640 medium containing heat inactivated 10% FBS, 1% penicillin and streptomycin. Cells were maintained at 37°C in a 5% CO2 incubator. These cells joined Taxol, DDP, Adr 2 µg/ml, respectively.
Experimental groups
The test was divided into six different concentration groups: normal group (no morusin), 1.25, 2.5, 5, 10 and 20 µg/mL (these concentrations are final concentrations).
MTT & CCK8 assay. The logarithmic growth cells were collected, and the appropriate concentration of cell suspension was selected. After each hole in the 96-well plate was added to 90 µl of cell suspension, it was cultivated overnight at 37°C in a 5% CO2 Incubator (HERACELL 150i type of Thermo Scientific). When cells were in the adherent condition, the control (normal) group and the morusin (sample) groups were respectively set up, each group with 4 compound holes, with 10 µl of morusin added to each experimental well and 10 µl of complete medium added to the control group. Then, they were cultured in the incubator for 24 h, 48 h and 72 h, added 20 µl MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 5mg/ml, Sigma) solution or 10 µl CCK (Cell Counting Kit 8, Nanjing KeyGen BioTech Co., Ltd., China) solution and cultivated for an additional 4 h. Finally, the culture solution was removed, and formazan crystals were dissolved in 150 µL of DMSO (dimethyl sulfoxide). Absorbance was then recorded at a 450 nm or 495 nm wavelength by a spectrophotometer (SpectraMax M5, Molecular Devices Company, USA). Tumour inhibitory rate and cell viability were calculated as follows:
Tumour inhibitory rate (%) = [1-A (morusin) / A (control)] Í 100%
Cell viability (%) = [A (morusin) – A (control)] / [A (no morusin) – A (control)] Í 100%
A (morusin): the absorbance of the wells containing CCK solution, cells and different doses of morusin
A (control): the absorbance of the wells containing CCK solution, complete medium and no cells
A (no morusin): the absorbance of the wells containing CCK solution, cells and no morusin
Cell apoptosis and cell cycle
The logarithmic growth cells were added to a 6-well plate in an incubator overnight. The morusin was added to the corresponding drug medium, and the control group was set. After cells were mixed with morusin for 48 h, the cells were collected with 0.25% trypsin enzyme [without ethylenediaminetetraacetic acid (EDTA)].
- Cells were centrifuged for 5 min, and the supernatant was abandoned and the precipitate was resuspended by complete medium. The 100 µl of MUSETM Annexin & Dead Cell Kit (Nanjing Fcmacs Biotech Co., Ltd., China) reagents were loaded into 1.5 ml centrifuge tubes, and 100 µl of cell suspension was shifted to each tube, which was slightly mixed and incubated for 20 min at room temperature, and the cell apoptosis was measured by MUSETM flow cytometry (Becton-Dickinson, San Jose, CA).
- Cell cycle immobilisation: cell pellets were collected by centrifuge and resuspended in PBS (phosphate-buffered saline) with 0. 2 mL of ice-cold 70% ethanol and then kept at -20°C overnight. Immobilised cells were centrifuged for 5 min at 300×g speed, and cleaned in 1×PBS, then resuspended by 200 µL of MUSETM Cell Cycle Kit (Nanjing Fcmacs Biotech Co., Ltd., China) reagents and incubated for 30 min at room temperature. Cell apoptosis was measured by MUSETM Flow Cytometry (Becton-Dickinson, San Jose, CA, USA).
Hoechst staining
- The ordinary cover glass was soaked in 70% ethanol for 5 min or longer, washed in PBS times, and then washed with the cell culture medium. The control group and morusin groups were set up, and the cover glass was placed into the 6-well plate, which was approximately 50 to 80% full, and the cells were cultivated overnight.
- Corresponding concentrations of morusin (including 5, 10 and 20 µg/ml) and complete medium were combined in morusin group wells and blank control group well. After cultivation for 48 h, the culture solution was discarded and 0.5 mL of fixed liquid (4% formaldehyde solution) was added and fixed for 15 min.
- The fixed liquid was discarded, the cells were washed twice (6 min) with 4°C precooling PBS, and then all liquid was absorbed.
- Hoechst 33258 staining solution (0.5 mL) was added to each hole and then dyed for 15 min at room temperature in the dark.
- The staining liquid was discarded, and the cells were washed twice (10 min) with 4°C precooling PBS.
- A drop of anti-fluorescence quenching sealant was placed on the glass slide, covered with the cell of cover glass, avoiding the bubble as much as possible.
- Ultraviolet excitation was used to observe and photograph the cells under the inverted fluorescence microscope.
Western Blotting
The expression of caspase-3, cleaved caspase-3, E-cadherin, N-cadherin, Vimentin, Bax, Bcl-2 and b-tubulin in samples and in cells irradiated by 6 MV x-ray in different groups were detected by WB. Total proteins from cells were centrifuged for 20 min at 13,000´g. Proteins were then shifted to polyvinylidene difluoride membranes (Millipore, Shanghai, China), closed with 5% non-fat milk in tris-buffered saline with Tween (TBST, 20 mM Tris–HCl, 150 mM NaCl, 0.05% Tween 20, pH 8.0) and buffered for 1 h. The target proteins were detected with different antibodies at 4°C overnight, and washed 4 times with TBST for 30 min. Then, the secondary antibodies were bioconjugated with horseradish peroxidase at a dilution (1:10000) for 1 h at room temperature and washed 4 times with TBST. The β-tubulin was used as the internal reference. The enhanced chemiluminescence (ECL) method was applied for development, and cassette exposure was used for development and fixation. Bands were visualised using a UVP detection system and band intensity that was quantified using LAB WORKS 4.6. All experimental results were repeated 3 times.
Statistical Analysis
The experimental data were recorded and analysed using Origin 8.5 software. The result as mean standard deviation (±SD) and analysis of variance (ANOVA) was used to evaluate the difference between multiple groups. SigmaScan Pro 5 software was used for WB light density determination, and X ±S (n = 3) expressed the final results, with P values less than 0.05 as significant and P values less than 0.01 as very significant.
{kind=link}